{"title":"MethylC-analyzer:用于全基因组DNA甲基化分析的综合下游管道。","authors":"Rita Jui-Hsien Lu, Pei-Yu Lin, Ming-Ren Yen, Bing-Heng Wu, Pao-Yang Chen","doi":"10.1186/s40529-022-00366-5","DOIUrl":null,"url":null,"abstract":"<p><p>DNA methylation is a crucial epigenetic modification involved in multiple biological processes and diseases. Current approaches for measuring genome-wide DNA methylation via bisulfite sequencing (BS-seq) include whole-genome bisulfite sequencing (WGBS), reduced representation bisulfite sequencing (RRBS), and enzymatic methyl-seq (EM-seq). The computational analysis tools available for BS-seq data include customized aligners for mapping bisulfite-converted reads and computational pipelines for downstream data analysis. Current post-alignment methylation tools are specialized for the interpretation of CG methylation, which is known to dominate mammalian genomes, however, non-CG methylation (CHG and CHH, where H refers to A, C, or T) is commonly observed in plants and fungi and is closely associated with gene regulation, transposon silencing, and plant development. Thus, we have developed a MethylC-analyzer to analyze and visualize post-alignment WGBS, RRBS, and EM-seq data focusing on CG. The tool is able to also analyze non-CG sites to enhance deciphering genomes of plants and fungi. By processing aligned data and gene location files, MethylC-analyzer generates a genome-wide view of methylation levels and methylation in user-specified genomic regions. The meta-plot, for example, allows the investigation of DNA methylation within specific genomic elements. Moreover, our tool identifies differentially methylated regions (DMRs) and investigates the enrichment of genomic features associated with variable methylation. MethylC-analyzer functionality is not limited to specific genomes, and we demonstrated its performance on both plant and human BS-seq data. MethylC-analyzer is a Python- and R-based program designed to perform comprehensive downstream analyses of methylation data, providing an intuitive analysis platform for scientists unfamiliar with DNA methylation analysis. It is available as either a standalone version for command-line uses or a graphical user interface (GUI) and is publicly accessible at https://github.com/RitataLU/MethylC-analyzer .</p>","PeriodicalId":9185,"journal":{"name":"Botanical Studies","volume":"64 1","pages":"1"},"PeriodicalIF":3.1000,"publicationDate":"2023-01-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9823188/pdf/","citationCount":"1","resultStr":"{\"title\":\"MethylC-analyzer: a comprehensive downstream pipeline for the analysis of genome-wide DNA methylation.\",\"authors\":\"Rita Jui-Hsien Lu, Pei-Yu Lin, Ming-Ren Yen, Bing-Heng Wu, Pao-Yang Chen\",\"doi\":\"10.1186/s40529-022-00366-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>DNA methylation is a crucial epigenetic modification involved in multiple biological processes and diseases. Current approaches for measuring genome-wide DNA methylation via bisulfite sequencing (BS-seq) include whole-genome bisulfite sequencing (WGBS), reduced representation bisulfite sequencing (RRBS), and enzymatic methyl-seq (EM-seq). The computational analysis tools available for BS-seq data include customized aligners for mapping bisulfite-converted reads and computational pipelines for downstream data analysis. Current post-alignment methylation tools are specialized for the interpretation of CG methylation, which is known to dominate mammalian genomes, however, non-CG methylation (CHG and CHH, where H refers to A, C, or T) is commonly observed in plants and fungi and is closely associated with gene regulation, transposon silencing, and plant development. Thus, we have developed a MethylC-analyzer to analyze and visualize post-alignment WGBS, RRBS, and EM-seq data focusing on CG. The tool is able to also analyze non-CG sites to enhance deciphering genomes of plants and fungi. By processing aligned data and gene location files, MethylC-analyzer generates a genome-wide view of methylation levels and methylation in user-specified genomic regions. The meta-plot, for example, allows the investigation of DNA methylation within specific genomic elements. Moreover, our tool identifies differentially methylated regions (DMRs) and investigates the enrichment of genomic features associated with variable methylation. MethylC-analyzer functionality is not limited to specific genomes, and we demonstrated its performance on both plant and human BS-seq data. MethylC-analyzer is a Python- and R-based program designed to perform comprehensive downstream analyses of methylation data, providing an intuitive analysis platform for scientists unfamiliar with DNA methylation analysis. It is available as either a standalone version for command-line uses or a graphical user interface (GUI) and is publicly accessible at https://github.com/RitataLU/MethylC-analyzer .</p>\",\"PeriodicalId\":9185,\"journal\":{\"name\":\"Botanical Studies\",\"volume\":\"64 1\",\"pages\":\"1\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2023-01-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9823188/pdf/\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Botanical Studies\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s40529-022-00366-5\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Agricultural and Biological Sciences\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Botanical Studies","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s40529-022-00366-5","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
MethylC-analyzer: a comprehensive downstream pipeline for the analysis of genome-wide DNA methylation.
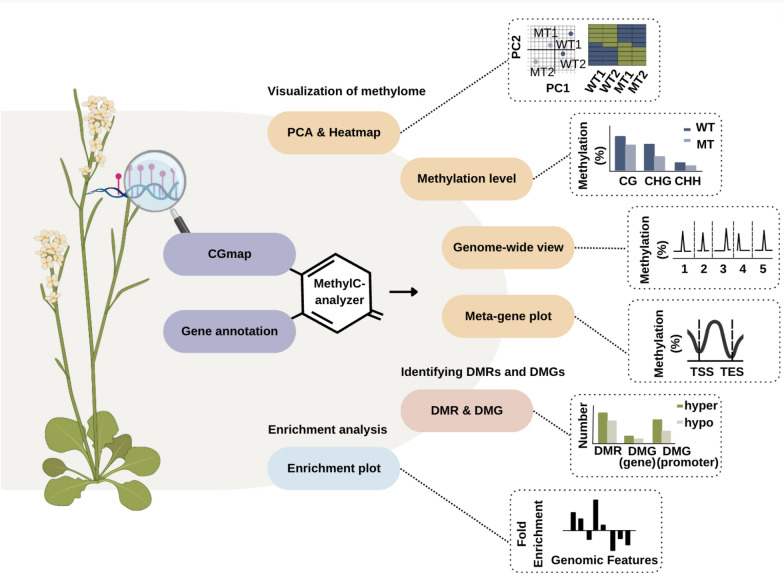
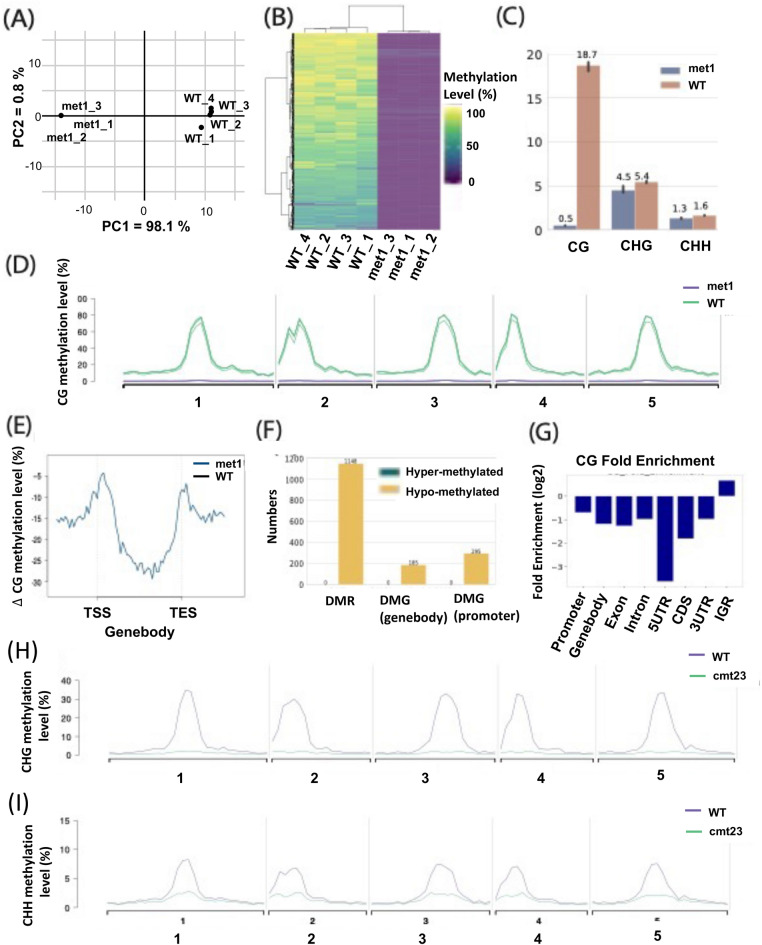
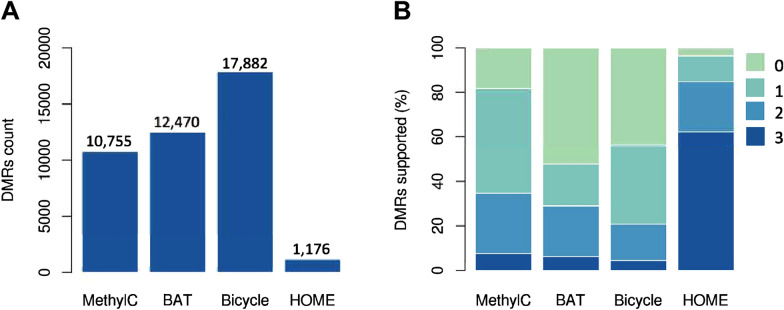
DNA methylation is a crucial epigenetic modification involved in multiple biological processes and diseases. Current approaches for measuring genome-wide DNA methylation via bisulfite sequencing (BS-seq) include whole-genome bisulfite sequencing (WGBS), reduced representation bisulfite sequencing (RRBS), and enzymatic methyl-seq (EM-seq). The computational analysis tools available for BS-seq data include customized aligners for mapping bisulfite-converted reads and computational pipelines for downstream data analysis. Current post-alignment methylation tools are specialized for the interpretation of CG methylation, which is known to dominate mammalian genomes, however, non-CG methylation (CHG and CHH, where H refers to A, C, or T) is commonly observed in plants and fungi and is closely associated with gene regulation, transposon silencing, and plant development. Thus, we have developed a MethylC-analyzer to analyze and visualize post-alignment WGBS, RRBS, and EM-seq data focusing on CG. The tool is able to also analyze non-CG sites to enhance deciphering genomes of plants and fungi. By processing aligned data and gene location files, MethylC-analyzer generates a genome-wide view of methylation levels and methylation in user-specified genomic regions. The meta-plot, for example, allows the investigation of DNA methylation within specific genomic elements. Moreover, our tool identifies differentially methylated regions (DMRs) and investigates the enrichment of genomic features associated with variable methylation. MethylC-analyzer functionality is not limited to specific genomes, and we demonstrated its performance on both plant and human BS-seq data. MethylC-analyzer is a Python- and R-based program designed to perform comprehensive downstream analyses of methylation data, providing an intuitive analysis platform for scientists unfamiliar with DNA methylation analysis. It is available as either a standalone version for command-line uses or a graphical user interface (GUI) and is publicly accessible at https://github.com/RitataLU/MethylC-analyzer .
期刊介绍:
Botanical Studies is an open access journal that encompasses all aspects of botany, including but not limited to taxonomy, morphology, development, genetics, evolution, reproduction, systematics, and biodiversity of all plant groups, algae, and fungi. The journal is affiliated with the Institute of Plant and Microbial Biology, Academia Sinica, Taiwan.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
