{"title":"假磷酸酶 STYXL1 的耗竭可通过 ER 压力增强葡萄糖脑苷脂向溶酶体的贩运。","authors":"Saloni Patel, Anshul Milap Bhatt, Priyanka Bhansali, Subba Rao Gangi Setty","doi":"10.1111/tra.12886","DOIUrl":null,"url":null,"abstract":"<p><p>Pseudophosphatases are catalytically inactive but share sequence and structural similarities with classical phosphatases. STYXL1 is a pseudophosphatase that belongs to the family of dual-specificity phosphatases and is known to regulate stress granule formation, neurite formation and apoptosis in different cell types. However, the role of STYXL1 in regulating cellular trafficking or the lysosome function has not been elucidated. Here, we show that the knockdown of STYXL1 enhances the trafficking of β-glucocerebrosidase (β-GC) and its lysosomal activity in HeLa cells. Importantly, the STYXL1-depleted cells display enhanced distribution of endoplasmic reticulum (ER), late endosome and lysosome compartments. Further, knockdown of STYXL1 causes the nuclear translocation of unfolded protein response (UPR) and lysosomal biogenesis transcription factors. However, the upregulated β-GC activity in the lysosomes is independent of TFEB/TFE3 nuclear localization in STYXL1 knockdown cells. The treatment of STYXL1 knockdown cells with 4-PBA (ER stress attenuator) significantly reduces the β-GC activity equivalent to control cells but not additive with thapsigargin, an ER stress activator. Additionally, STYXL1-depleted cells show the enhanced contact of lysosomes with ER, possibly via increased UPR. The depletion of STYXL1 in human primary fibroblasts derived from Gaucher patients showed moderately enhanced lysosomal enzyme activity. Overall, these studies illustrated the unique role of pseudophosphatase STYXL1 in modulating the lysosome function both in normal and lysosome-storage disorder cell types. Thus, designing small molecules against STYXL1 possibly can restore the lysosome activity by enhancing ER stress in Gaucher disease.</p>","PeriodicalId":23207,"journal":{"name":"Traffic","volume":"24 7","pages":"254-269"},"PeriodicalIF":2.5000,"publicationDate":"2023-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Pseudophosphatase STYXL1 depletion enhances glucocerebrosidase trafficking to lysosomes via ER stress.\",\"authors\":\"Saloni Patel, Anshul Milap Bhatt, Priyanka Bhansali, Subba Rao Gangi Setty\",\"doi\":\"10.1111/tra.12886\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Pseudophosphatases are catalytically inactive but share sequence and structural similarities with classical phosphatases. STYXL1 is a pseudophosphatase that belongs to the family of dual-specificity phosphatases and is known to regulate stress granule formation, neurite formation and apoptosis in different cell types. However, the role of STYXL1 in regulating cellular trafficking or the lysosome function has not been elucidated. Here, we show that the knockdown of STYXL1 enhances the trafficking of β-glucocerebrosidase (β-GC) and its lysosomal activity in HeLa cells. Importantly, the STYXL1-depleted cells display enhanced distribution of endoplasmic reticulum (ER), late endosome and lysosome compartments. Further, knockdown of STYXL1 causes the nuclear translocation of unfolded protein response (UPR) and lysosomal biogenesis transcription factors. However, the upregulated β-GC activity in the lysosomes is independent of TFEB/TFE3 nuclear localization in STYXL1 knockdown cells. The treatment of STYXL1 knockdown cells with 4-PBA (ER stress attenuator) significantly reduces the β-GC activity equivalent to control cells but not additive with thapsigargin, an ER stress activator. Additionally, STYXL1-depleted cells show the enhanced contact of lysosomes with ER, possibly via increased UPR. The depletion of STYXL1 in human primary fibroblasts derived from Gaucher patients showed moderately enhanced lysosomal enzyme activity. Overall, these studies illustrated the unique role of pseudophosphatase STYXL1 in modulating the lysosome function both in normal and lysosome-storage disorder cell types. Thus, designing small molecules against STYXL1 possibly can restore the lysosome activity by enhancing ER stress in Gaucher disease.</p>\",\"PeriodicalId\":23207,\"journal\":{\"name\":\"Traffic\",\"volume\":\"24 7\",\"pages\":\"254-269\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2023-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Traffic\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1111/tra.12886\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2023/5/17 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Traffic","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1111/tra.12886","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2023/5/17 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
Pseudophosphatase STYXL1 depletion enhances glucocerebrosidase trafficking to lysosomes via ER stress.
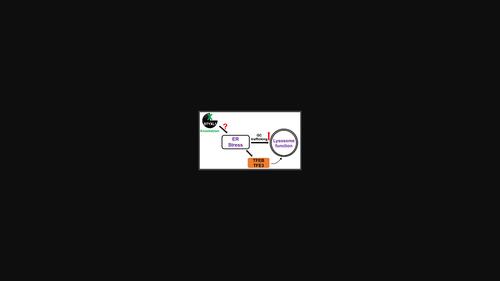
Pseudophosphatases are catalytically inactive but share sequence and structural similarities with classical phosphatases. STYXL1 is a pseudophosphatase that belongs to the family of dual-specificity phosphatases and is known to regulate stress granule formation, neurite formation and apoptosis in different cell types. However, the role of STYXL1 in regulating cellular trafficking or the lysosome function has not been elucidated. Here, we show that the knockdown of STYXL1 enhances the trafficking of β-glucocerebrosidase (β-GC) and its lysosomal activity in HeLa cells. Importantly, the STYXL1-depleted cells display enhanced distribution of endoplasmic reticulum (ER), late endosome and lysosome compartments. Further, knockdown of STYXL1 causes the nuclear translocation of unfolded protein response (UPR) and lysosomal biogenesis transcription factors. However, the upregulated β-GC activity in the lysosomes is independent of TFEB/TFE3 nuclear localization in STYXL1 knockdown cells. The treatment of STYXL1 knockdown cells with 4-PBA (ER stress attenuator) significantly reduces the β-GC activity equivalent to control cells but not additive with thapsigargin, an ER stress activator. Additionally, STYXL1-depleted cells show the enhanced contact of lysosomes with ER, possibly via increased UPR. The depletion of STYXL1 in human primary fibroblasts derived from Gaucher patients showed moderately enhanced lysosomal enzyme activity. Overall, these studies illustrated the unique role of pseudophosphatase STYXL1 in modulating the lysosome function both in normal and lysosome-storage disorder cell types. Thus, designing small molecules against STYXL1 possibly can restore the lysosome activity by enhancing ER stress in Gaucher disease.
期刊介绍:
Traffic encourages and facilitates the publication of papers in any field relating to intracellular transport in health and disease. Traffic papers span disciplines such as developmental biology, neuroscience, innate and adaptive immunity, epithelial cell biology, intracellular pathogens and host-pathogen interactions, among others using any eukaryotic model system. Areas of particular interest include protein, nucleic acid and lipid traffic, molecular motors, intracellular pathogens, intracellular proteolysis, nuclear import and export, cytokinesis and the cell cycle, the interface between signaling and trafficking or localization, protein translocation, the cell biology of adaptive an innate immunity, organelle biogenesis, metabolism, cell polarity and organization, and organelle movement.
All aspects of the structural, molecular biology, biochemistry, genetics, morphology, intracellular signaling and relationship to hereditary or infectious diseases will be covered. Manuscripts must provide a clear conceptual or mechanistic advance. The editors will reject papers that require major changes, including addition of significant experimental data or other significant revision.
Traffic will consider manuscripts of any length, but encourages authors to limit their papers to 16 typeset pages or less.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
