Yvonne Hort, Patricia Sullivan, Laura Wedd, Lindsay Fowles, Igor Stevanovski, Ira Deveson, Cas Simons, Andrew Mallett, Chirag Patel, Timothy Furlong, Mark J Cowley, John Shine, Amali Mallawaarachchi
{"title":"PKD1的非典型剪接变异解释了大多数未确诊的典型家族性ADPKD。","authors":"Yvonne Hort, Patricia Sullivan, Laura Wedd, Lindsay Fowles, Igor Stevanovski, Ira Deveson, Cas Simons, Andrew Mallett, Chirag Patel, Timothy Furlong, Mark J Cowley, John Shine, Amali Mallawaarachchi","doi":"10.1038/s41525-023-00362-z","DOIUrl":null,"url":null,"abstract":"<p><p>Autosomal dominant polycystic kidney disease (ADPKD) is the most common monogenic cause of kidney failure and is primarily associated with PKD1 or PKD2. Approximately 10% of patients remain undiagnosed after standard genetic testing. We aimed to utilise short and long-read genome sequencing and RNA studies to investigate undiagnosed families. Patients with typical ADPKD phenotype and undiagnosed after genetic diagnostics were recruited. Probands underwent short-read genome sequencing, PKD1 and PKD2 coding and non-coding analyses and then genome-wide analysis. Targeted RNA studies investigated variants suspected to impact splicing. Those undiagnosed then underwent Oxford Nanopore Technologies long-read genome sequencing. From over 172 probands, 9 met inclusion criteria and consented. A genetic diagnosis was made in 8 of 9 (89%) families undiagnosed on prior genetic testing. Six had variants impacting splicing, five in non-coding regions of PKD1. Short-read genome sequencing identified novel branchpoint, AG-exclusion zone and missense variants generating cryptic splice sites and a deletion causing critical intron shortening. Long-read sequencing confirmed the diagnosis in one family. Most undiagnosed families with typical ADPKD have splice-impacting variants in PKD1. We describe a pragmatic method for diagnostic laboratories to assess PKD1 and PKD2 non-coding regions and validate suspected splicing variants through targeted RNA studies.</p>","PeriodicalId":19273,"journal":{"name":"NPJ Genomic Medicine","volume":"8 1","pages":"16"},"PeriodicalIF":4.8000,"publicationDate":"2023-07-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10328916/pdf/","citationCount":"0","resultStr":"{\"title\":\"Atypical splicing variants in PKD1 explain most undiagnosed typical familial ADPKD.\",\"authors\":\"Yvonne Hort, Patricia Sullivan, Laura Wedd, Lindsay Fowles, Igor Stevanovski, Ira Deveson, Cas Simons, Andrew Mallett, Chirag Patel, Timothy Furlong, Mark J Cowley, John Shine, Amali Mallawaarachchi\",\"doi\":\"10.1038/s41525-023-00362-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Autosomal dominant polycystic kidney disease (ADPKD) is the most common monogenic cause of kidney failure and is primarily associated with PKD1 or PKD2. Approximately 10% of patients remain undiagnosed after standard genetic testing. We aimed to utilise short and long-read genome sequencing and RNA studies to investigate undiagnosed families. Patients with typical ADPKD phenotype and undiagnosed after genetic diagnostics were recruited. Probands underwent short-read genome sequencing, PKD1 and PKD2 coding and non-coding analyses and then genome-wide analysis. Targeted RNA studies investigated variants suspected to impact splicing. Those undiagnosed then underwent Oxford Nanopore Technologies long-read genome sequencing. From over 172 probands, 9 met inclusion criteria and consented. A genetic diagnosis was made in 8 of 9 (89%) families undiagnosed on prior genetic testing. Six had variants impacting splicing, five in non-coding regions of PKD1. Short-read genome sequencing identified novel branchpoint, AG-exclusion zone and missense variants generating cryptic splice sites and a deletion causing critical intron shortening. Long-read sequencing confirmed the diagnosis in one family. Most undiagnosed families with typical ADPKD have splice-impacting variants in PKD1. We describe a pragmatic method for diagnostic laboratories to assess PKD1 and PKD2 non-coding regions and validate suspected splicing variants through targeted RNA studies.</p>\",\"PeriodicalId\":19273,\"journal\":{\"name\":\"NPJ Genomic Medicine\",\"volume\":\"8 1\",\"pages\":\"16\"},\"PeriodicalIF\":4.8000,\"publicationDate\":\"2023-07-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10328916/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NPJ Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41525-023-00362-z\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41525-023-00362-z","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Atypical splicing variants in PKD1 explain most undiagnosed typical familial ADPKD.
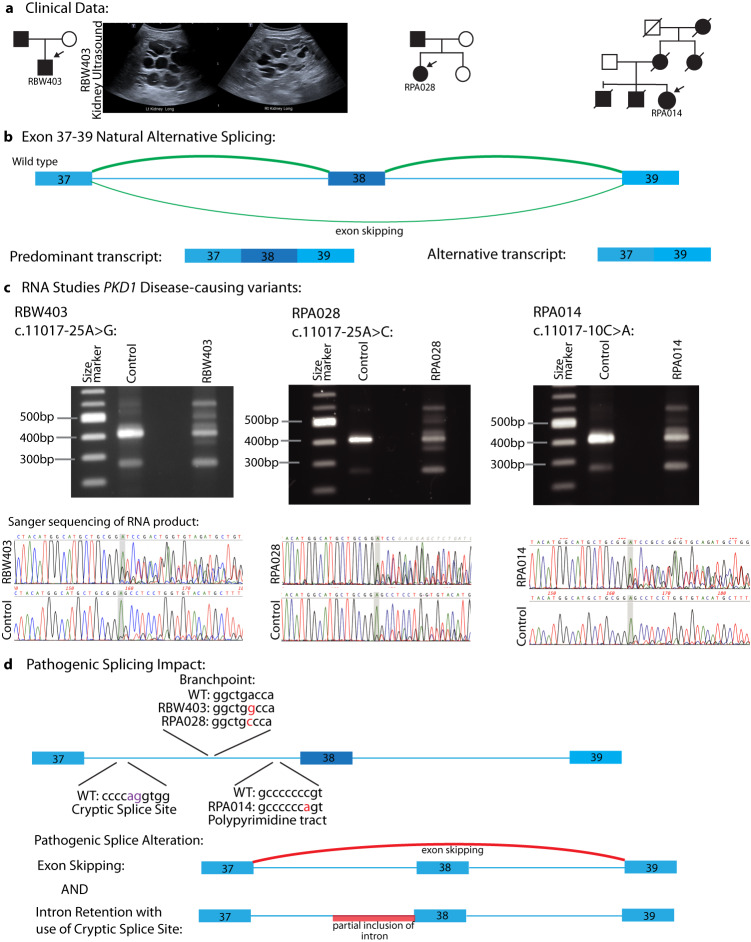
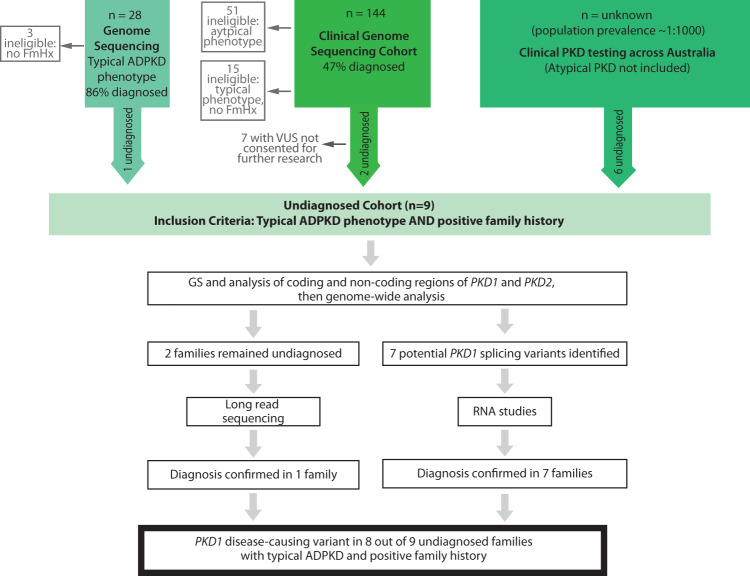
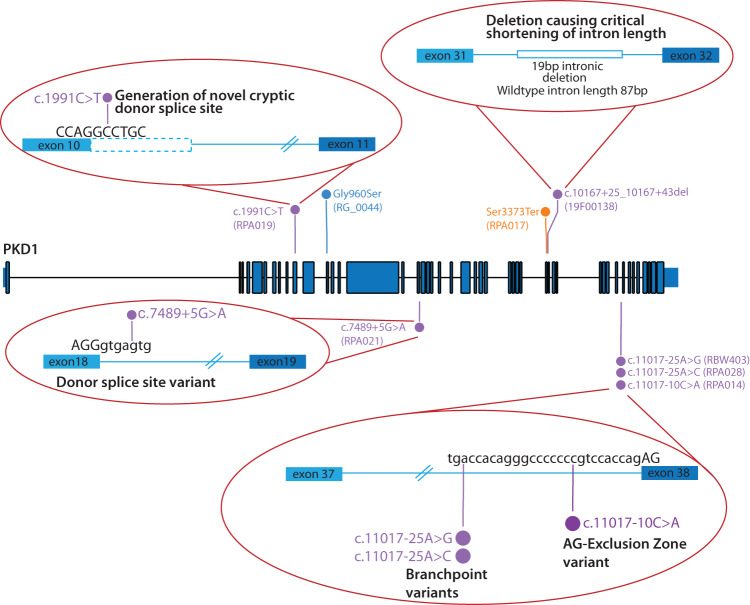
Autosomal dominant polycystic kidney disease (ADPKD) is the most common monogenic cause of kidney failure and is primarily associated with PKD1 or PKD2. Approximately 10% of patients remain undiagnosed after standard genetic testing. We aimed to utilise short and long-read genome sequencing and RNA studies to investigate undiagnosed families. Patients with typical ADPKD phenotype and undiagnosed after genetic diagnostics were recruited. Probands underwent short-read genome sequencing, PKD1 and PKD2 coding and non-coding analyses and then genome-wide analysis. Targeted RNA studies investigated variants suspected to impact splicing. Those undiagnosed then underwent Oxford Nanopore Technologies long-read genome sequencing. From over 172 probands, 9 met inclusion criteria and consented. A genetic diagnosis was made in 8 of 9 (89%) families undiagnosed on prior genetic testing. Six had variants impacting splicing, five in non-coding regions of PKD1. Short-read genome sequencing identified novel branchpoint, AG-exclusion zone and missense variants generating cryptic splice sites and a deletion causing critical intron shortening. Long-read sequencing confirmed the diagnosis in one family. Most undiagnosed families with typical ADPKD have splice-impacting variants in PKD1. We describe a pragmatic method for diagnostic laboratories to assess PKD1 and PKD2 non-coding regions and validate suspected splicing variants through targeted RNA studies.
NPJ Genomic MedicineBiochemistry, Genetics and Molecular Biology-Molecular Biology
CiteScore
9.40
自引率
1.90%
发文量
67
审稿时长
17 weeks
期刊介绍:
npj Genomic Medicine is an international, peer-reviewed journal dedicated to publishing the most important scientific advances in all aspects of genomics and its application in the practice of medicine.
The journal defines genomic medicine as "diagnosis, prognosis, prevention and/or treatment of disease and disorders of the mind and body, using approaches informed or enabled by knowledge of the genome and the molecules it encodes." Relevant and high-impact papers that encompass studies of individuals, families, or populations are considered for publication. An emphasis will include coupling detailed phenotype and genome sequencing information, both enabled by new technologies and informatics, to delineate the underlying aetiology of disease. Clinical recommendations and/or guidelines of how that data should be used in the clinical management of those patients in the study, and others, are also encouraged.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
