Zicheng Zhang, Hongyan Chen, Dongxue Yan, Lu Chen, Jie Sun, Meng Zhou
{"title":"深度学习识别t细胞耗竭依赖的转录特征,用于预测临床结果和对免疫检查点封锁的反应。","authors":"Zicheng Zhang, Hongyan Chen, Dongxue Yan, Lu Chen, Jie Sun, Meng Zhou","doi":"10.1038/s41389-023-00482-2","DOIUrl":null,"url":null,"abstract":"<p><p>Immune checkpoint blockade (ICB) therapies have brought unprecedented advances in cancer treatment, but responses are limited to a fraction of patients. Therefore, sustained and substantial efforts are required to advance clinical and translational investigation on managing patients receiving ICB. In this study, we investigated the dynamic changes in molecular profiles of T-cell exhaustion (TEX) during ICB treatment using single-cell and bulk transcriptome analysis, and demonstrated distinct exhaustion molecular profiles associated with ICB response. By applying an ensemble deep-learning computational framework, we identified an ICB-associated transcriptional signature consisting of 16 TEX-related genes, termed ITGs. Incorporating 16 ITGs into a machine-learning model called MLTIP achieved reliable predictive power for clinical ICB response with an average AUC of 0.778, and overall survival (pooled HR = 0.093, 95% CI, 0.031-0.28, P < 0.001) across multiple ICB-treated cohorts. Furthermore, the MLTIP consistently demonstrated superior predictive performance compared to other well-established markers and signatures, with an average increase in AUC of 21.5%. In summary, our results highlight the potential of this TEX-dependent transcriptional signature as a tool for precise patient stratification and personalized immunotherapy, with clinical translation in precision medicine.</p>","PeriodicalId":19489,"journal":{"name":"Oncogenesis","volume":"12 1","pages":"37"},"PeriodicalIF":5.9000,"publicationDate":"2023-07-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10336094/pdf/","citationCount":"1","resultStr":"{\"title\":\"Deep learning identifies a T-cell exhaustion-dependent transcriptional signature for predicting clinical outcomes and response to immune checkpoint blockade.\",\"authors\":\"Zicheng Zhang, Hongyan Chen, Dongxue Yan, Lu Chen, Jie Sun, Meng Zhou\",\"doi\":\"10.1038/s41389-023-00482-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Immune checkpoint blockade (ICB) therapies have brought unprecedented advances in cancer treatment, but responses are limited to a fraction of patients. Therefore, sustained and substantial efforts are required to advance clinical and translational investigation on managing patients receiving ICB. In this study, we investigated the dynamic changes in molecular profiles of T-cell exhaustion (TEX) during ICB treatment using single-cell and bulk transcriptome analysis, and demonstrated distinct exhaustion molecular profiles associated with ICB response. By applying an ensemble deep-learning computational framework, we identified an ICB-associated transcriptional signature consisting of 16 TEX-related genes, termed ITGs. Incorporating 16 ITGs into a machine-learning model called MLTIP achieved reliable predictive power for clinical ICB response with an average AUC of 0.778, and overall survival (pooled HR = 0.093, 95% CI, 0.031-0.28, P < 0.001) across multiple ICB-treated cohorts. Furthermore, the MLTIP consistently demonstrated superior predictive performance compared to other well-established markers and signatures, with an average increase in AUC of 21.5%. In summary, our results highlight the potential of this TEX-dependent transcriptional signature as a tool for precise patient stratification and personalized immunotherapy, with clinical translation in precision medicine.</p>\",\"PeriodicalId\":19489,\"journal\":{\"name\":\"Oncogenesis\",\"volume\":\"12 1\",\"pages\":\"37\"},\"PeriodicalIF\":5.9000,\"publicationDate\":\"2023-07-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10336094/pdf/\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Oncogenesis\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41389-023-00482-2\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"ONCOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Oncogenesis","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41389-023-00482-2","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ONCOLOGY","Score":null,"Total":0}
引用次数: 1
摘要
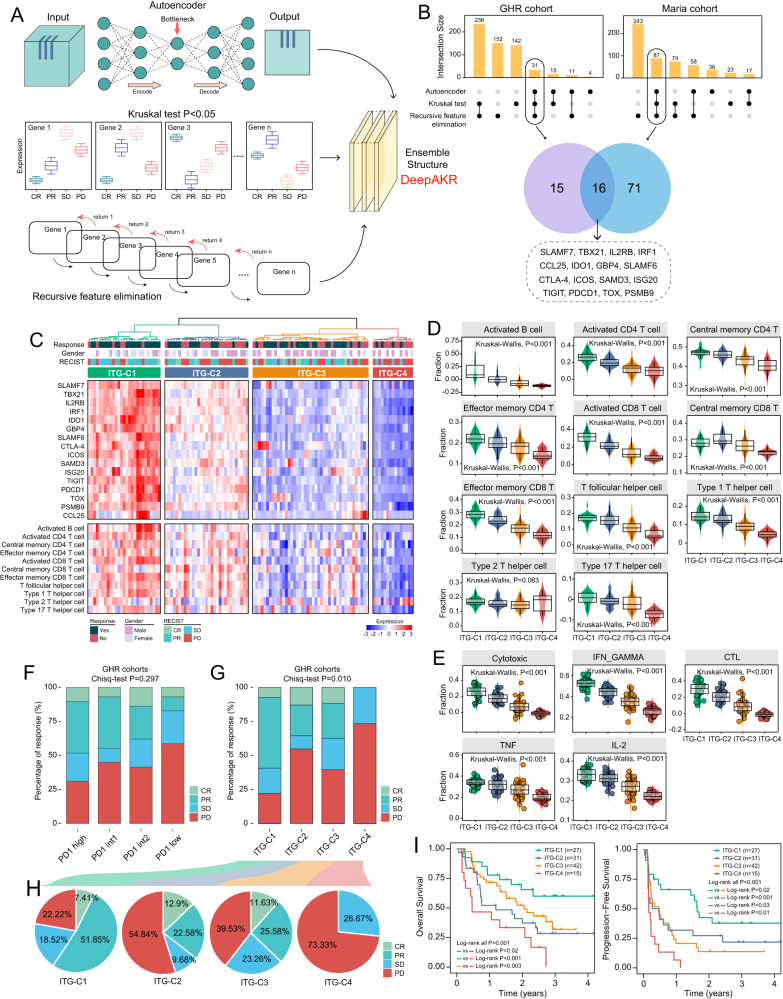
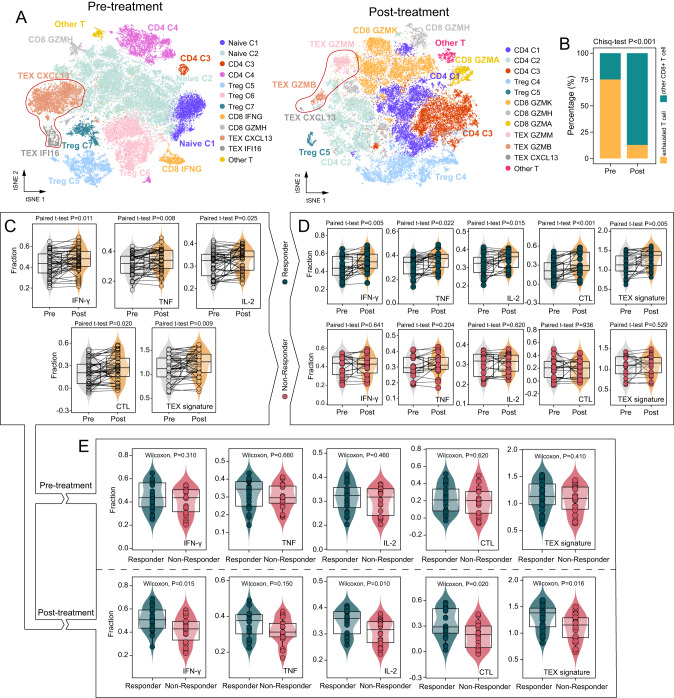
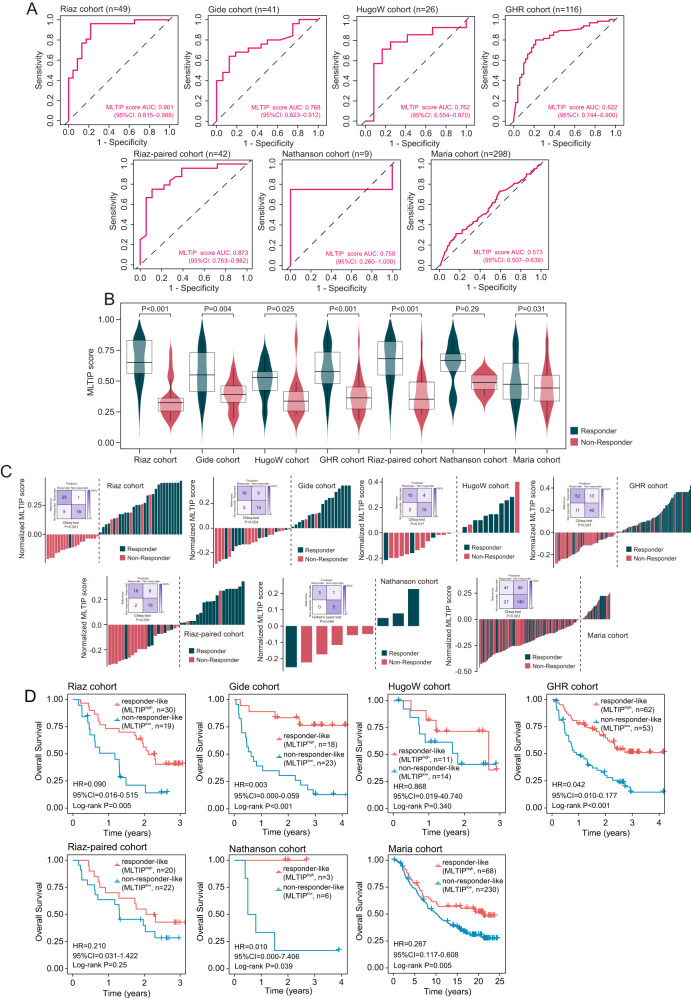
免疫检查点阻断(ICB)疗法在癌症治疗方面取得了前所未有的进展,但反应仅限于一小部分患者。因此,需要持续和大量的努力来推进临床和转化研究,以管理接受ICB的患者。在这项研究中,我们利用单细胞和大量转录组分析研究了ICB治疗期间t细胞衰竭(TEX)分子谱的动态变化,并证明了与ICB反应相关的不同的衰竭分子谱。通过应用集成深度学习计算框架,我们确定了由16个texg相关基因组成的icb相关转录特征,称为ITGs。将16个ITGs纳入名为MLTIP的机器学习模型中,获得了临床ICB反应的可靠预测能力,平均AUC为0.778,总生存期(合并HR = 0.093, 95% CI, 0.031-0.28, P
Deep learning identifies a T-cell exhaustion-dependent transcriptional signature for predicting clinical outcomes and response to immune checkpoint blockade.
Immune checkpoint blockade (ICB) therapies have brought unprecedented advances in cancer treatment, but responses are limited to a fraction of patients. Therefore, sustained and substantial efforts are required to advance clinical and translational investigation on managing patients receiving ICB. In this study, we investigated the dynamic changes in molecular profiles of T-cell exhaustion (TEX) during ICB treatment using single-cell and bulk transcriptome analysis, and demonstrated distinct exhaustion molecular profiles associated with ICB response. By applying an ensemble deep-learning computational framework, we identified an ICB-associated transcriptional signature consisting of 16 TEX-related genes, termed ITGs. Incorporating 16 ITGs into a machine-learning model called MLTIP achieved reliable predictive power for clinical ICB response with an average AUC of 0.778, and overall survival (pooled HR = 0.093, 95% CI, 0.031-0.28, P < 0.001) across multiple ICB-treated cohorts. Furthermore, the MLTIP consistently demonstrated superior predictive performance compared to other well-established markers and signatures, with an average increase in AUC of 21.5%. In summary, our results highlight the potential of this TEX-dependent transcriptional signature as a tool for precise patient stratification and personalized immunotherapy, with clinical translation in precision medicine.
期刊介绍:
Oncogenesis is a peer-reviewed open access online journal that publishes full-length papers, reviews, and short communications exploring the molecular basis of cancer and related phenomena. It seeks to promote diverse and integrated areas of molecular biology, cell biology, oncology, and genetics.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
