P Reventun, S Sánchez-Esteban, A Cook-Calvete, M Delgado-Marín, C Roza, S Jorquera-Ortega, I Hernandez, L Tesoro, L Botana, J L Zamorano, C Zaragoza, M Saura
{"title":"内皮ILK通过防止冠状动脉微血管功能障碍和内皮-间质转化诱导心脏保护。","authors":"P Reventun, S Sánchez-Esteban, A Cook-Calvete, M Delgado-Marín, C Roza, S Jorquera-Ortega, I Hernandez, L Tesoro, L Botana, J L Zamorano, C Zaragoza, M Saura","doi":"10.1007/s00395-023-00997-0","DOIUrl":null,"url":null,"abstract":"<p><p>Endothelial dysfunction is an early event in coronary microvascular disease. Integrin-linked kinase (ILK) prevents endothelial nitric oxide synthase (eNOS) uncoupling and, thus, endothelial dysfunction. However, the specific role of endothelial ILK in cardiac function remains to be fully elucidated. We hypothesised that endothelial ILK plays a crucial role in maintaining coronary microvascular function and contractile performance in the heart. We generated an endothelial cell-specific ILK conditional knock-out mouse (ecILK cKO) and investigated cardiovascular function. Coronary endothelial ILK deletion significantly impaired cardiac function: ejection fraction, fractional shortening and cardiac output decreased, whilst left ventricle diastolic internal diameter decreased and E/A and E/E' ratios increased, indicating not only systolic but also diastolic dysfunction. The functional data correlated with extensive extracellular matrix remodelling and perivascular fibrosis, indicative of adverse cardiac remodelling. Mice with endothelial ILK deletion suffered early ischaemic-like events with ST elevation and transient increases in cardiac troponins, which correlated with fibrotic remodelling. In addition, ecILK cKO mice exhibited many features of coronary microvascular disease: reduced cardiac perfusion, impaired coronary flow reserve and arterial remodelling with patent epicardial coronary arteries. Moreover, endothelial ILK deletion induced a moderate increase in blood pressure, but the antihypertensive drug Losartan did not affect microvascular remodelling whilst only partially ameliorated fibrotic remodelling. The plasma miRNA profile reveals endothelial-to-mesenchymal transition (endMT) as an upregulated pathway in endothelial ILK conditional KO mice. Our results show that endothelial cells in the microvasculature in endothelial ILK conditional KO mice underwent endMT. Moreover, endothelial cells isolated from these mice and ILK-silenced human microvascular endothelial cells underwent endMT, indicating that decreased endothelial ILK contributes directly to this endothelial phenotype shift. Our results identify ILK as a crucial regulator of microvascular endothelial homeostasis. Endothelial ILK prevents microvascular dysfunction and cardiac remodelling, contributing to the maintenance of the endothelial cell phenotype.</p>","PeriodicalId":8723,"journal":{"name":"Basic Research in Cardiology","volume":"118 1","pages":"28"},"PeriodicalIF":8.0000,"publicationDate":"2023-07-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10348984/pdf/","citationCount":"0","resultStr":"{\"title\":\"Endothelial ILK induces cardioprotection by preventing coronary microvascular dysfunction and endothelial-to-mesenchymal transition.\",\"authors\":\"P Reventun, S Sánchez-Esteban, A Cook-Calvete, M Delgado-Marín, C Roza, S Jorquera-Ortega, I Hernandez, L Tesoro, L Botana, J L Zamorano, C Zaragoza, M Saura\",\"doi\":\"10.1007/s00395-023-00997-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Endothelial dysfunction is an early event in coronary microvascular disease. Integrin-linked kinase (ILK) prevents endothelial nitric oxide synthase (eNOS) uncoupling and, thus, endothelial dysfunction. However, the specific role of endothelial ILK in cardiac function remains to be fully elucidated. We hypothesised that endothelial ILK plays a crucial role in maintaining coronary microvascular function and contractile performance in the heart. We generated an endothelial cell-specific ILK conditional knock-out mouse (ecILK cKO) and investigated cardiovascular function. Coronary endothelial ILK deletion significantly impaired cardiac function: ejection fraction, fractional shortening and cardiac output decreased, whilst left ventricle diastolic internal diameter decreased and E/A and E/E' ratios increased, indicating not only systolic but also diastolic dysfunction. The functional data correlated with extensive extracellular matrix remodelling and perivascular fibrosis, indicative of adverse cardiac remodelling. Mice with endothelial ILK deletion suffered early ischaemic-like events with ST elevation and transient increases in cardiac troponins, which correlated with fibrotic remodelling. In addition, ecILK cKO mice exhibited many features of coronary microvascular disease: reduced cardiac perfusion, impaired coronary flow reserve and arterial remodelling with patent epicardial coronary arteries. Moreover, endothelial ILK deletion induced a moderate increase in blood pressure, but the antihypertensive drug Losartan did not affect microvascular remodelling whilst only partially ameliorated fibrotic remodelling. The plasma miRNA profile reveals endothelial-to-mesenchymal transition (endMT) as an upregulated pathway in endothelial ILK conditional KO mice. Our results show that endothelial cells in the microvasculature in endothelial ILK conditional KO mice underwent endMT. Moreover, endothelial cells isolated from these mice and ILK-silenced human microvascular endothelial cells underwent endMT, indicating that decreased endothelial ILK contributes directly to this endothelial phenotype shift. Our results identify ILK as a crucial regulator of microvascular endothelial homeostasis. Endothelial ILK prevents microvascular dysfunction and cardiac remodelling, contributing to the maintenance of the endothelial cell phenotype.</p>\",\"PeriodicalId\":8723,\"journal\":{\"name\":\"Basic Research in Cardiology\",\"volume\":\"118 1\",\"pages\":\"28\"},\"PeriodicalIF\":8.0000,\"publicationDate\":\"2023-07-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10348984/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Basic Research in Cardiology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s00395-023-00997-0\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CARDIAC & CARDIOVASCULAR SYSTEMS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Basic Research in Cardiology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s00395-023-00997-0","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CARDIAC & CARDIOVASCULAR SYSTEMS","Score":null,"Total":0}
Endothelial ILK induces cardioprotection by preventing coronary microvascular dysfunction and endothelial-to-mesenchymal transition.
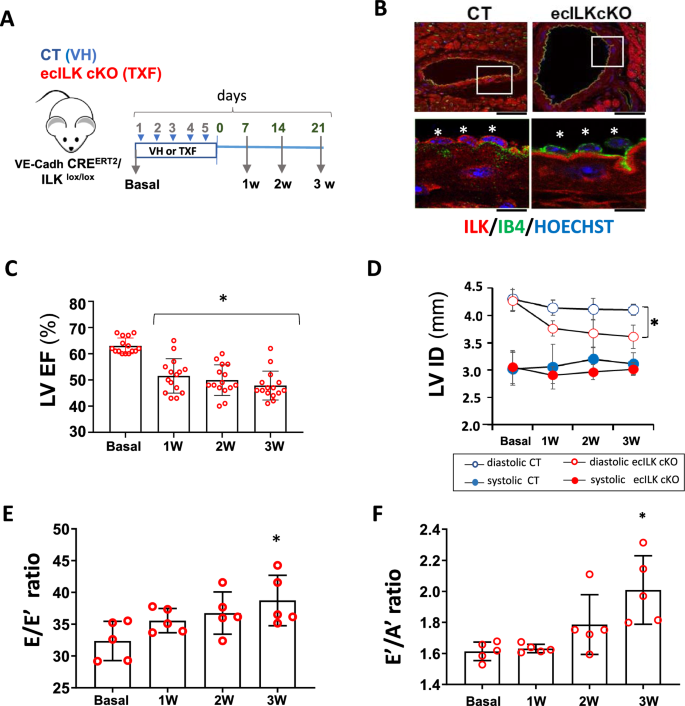
Endothelial dysfunction is an early event in coronary microvascular disease. Integrin-linked kinase (ILK) prevents endothelial nitric oxide synthase (eNOS) uncoupling and, thus, endothelial dysfunction. However, the specific role of endothelial ILK in cardiac function remains to be fully elucidated. We hypothesised that endothelial ILK plays a crucial role in maintaining coronary microvascular function and contractile performance in the heart. We generated an endothelial cell-specific ILK conditional knock-out mouse (ecILK cKO) and investigated cardiovascular function. Coronary endothelial ILK deletion significantly impaired cardiac function: ejection fraction, fractional shortening and cardiac output decreased, whilst left ventricle diastolic internal diameter decreased and E/A and E/E' ratios increased, indicating not only systolic but also diastolic dysfunction. The functional data correlated with extensive extracellular matrix remodelling and perivascular fibrosis, indicative of adverse cardiac remodelling. Mice with endothelial ILK deletion suffered early ischaemic-like events with ST elevation and transient increases in cardiac troponins, which correlated with fibrotic remodelling. In addition, ecILK cKO mice exhibited many features of coronary microvascular disease: reduced cardiac perfusion, impaired coronary flow reserve and arterial remodelling with patent epicardial coronary arteries. Moreover, endothelial ILK deletion induced a moderate increase in blood pressure, but the antihypertensive drug Losartan did not affect microvascular remodelling whilst only partially ameliorated fibrotic remodelling. The plasma miRNA profile reveals endothelial-to-mesenchymal transition (endMT) as an upregulated pathway in endothelial ILK conditional KO mice. Our results show that endothelial cells in the microvasculature in endothelial ILK conditional KO mice underwent endMT. Moreover, endothelial cells isolated from these mice and ILK-silenced human microvascular endothelial cells underwent endMT, indicating that decreased endothelial ILK contributes directly to this endothelial phenotype shift. Our results identify ILK as a crucial regulator of microvascular endothelial homeostasis. Endothelial ILK prevents microvascular dysfunction and cardiac remodelling, contributing to the maintenance of the endothelial cell phenotype.
期刊介绍:
Basic Research in Cardiology is an international journal for cardiovascular research. It provides a forum for original and review articles related to experimental cardiology that meet its stringent scientific standards.
Basic Research in Cardiology regularly receives articles from the fields of
- Molecular and Cellular Biology
- Biochemistry
- Biophysics
- Pharmacology
- Physiology and Pathology
- Clinical Cardiology



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
