Dinglin Zhang, Lidong Gong, Junben Weng, Yan Li, Anhui Wang, Guohui Li
{"title":"基于5微珠模型和多尺度模拟的RNA折叠。","authors":"Dinglin Zhang, Lidong Gong, Junben Weng, Yan Li, Anhui Wang, Guohui Li","doi":"10.1007/s12539-023-00561-3","DOIUrl":null,"url":null,"abstract":"<p><p>RNA folding prediction is very meaningful and challenging. The molecular dynamics simulation (MDS) of all atoms (AA) is limited to the folding of small RNA molecules. At present, most of the practical models are coarse grained (CG) model, and the coarse-grained force field (CGFF) parameters usually depend on known RNA structures. However, the limitation of the CGFF is obvious that it is difficult to study the modified RNA. Based on the 3 beads model (AIMS_RNA_B3), we proposed the AIMS_RNA_B5 model with three beads representing a base and two beads representing the main chain (sugar group and phosphate group). We first run the all atom molecular dynamic simulation (AAMDS), and fit the CGFF parameter with the AA trajectory. Then perform the coarse-grained molecular dynamic simulation (CGMDS). AAMDS is the foundation of CGMDS. CGMDS is mainly to carry out the conformation sampling based on the current AAMDS state and improve the folding speed. We simulated the folding of three RNAs, which belong to hairpin, pseudoknot and tRNA respectively. Compared to the AIMS_RNA_B3 model, the AIMS_RNA_B5 model is more reasonable and performs better.</p>","PeriodicalId":13670,"journal":{"name":"Interdisciplinary Sciences: Computational Life Sciences","volume":"15 3","pages":"393-404"},"PeriodicalIF":3.9000,"publicationDate":"2023-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"1","resultStr":"{\"title\":\"RNA Folding Based on 5 Beads Model and Multiscale Simulation.\",\"authors\":\"Dinglin Zhang, Lidong Gong, Junben Weng, Yan Li, Anhui Wang, Guohui Li\",\"doi\":\"10.1007/s12539-023-00561-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>RNA folding prediction is very meaningful and challenging. The molecular dynamics simulation (MDS) of all atoms (AA) is limited to the folding of small RNA molecules. At present, most of the practical models are coarse grained (CG) model, and the coarse-grained force field (CGFF) parameters usually depend on known RNA structures. However, the limitation of the CGFF is obvious that it is difficult to study the modified RNA. Based on the 3 beads model (AIMS_RNA_B3), we proposed the AIMS_RNA_B5 model with three beads representing a base and two beads representing the main chain (sugar group and phosphate group). We first run the all atom molecular dynamic simulation (AAMDS), and fit the CGFF parameter with the AA trajectory. Then perform the coarse-grained molecular dynamic simulation (CGMDS). AAMDS is the foundation of CGMDS. CGMDS is mainly to carry out the conformation sampling based on the current AAMDS state and improve the folding speed. We simulated the folding of three RNAs, which belong to hairpin, pseudoknot and tRNA respectively. Compared to the AIMS_RNA_B3 model, the AIMS_RNA_B5 model is more reasonable and performs better.</p>\",\"PeriodicalId\":13670,\"journal\":{\"name\":\"Interdisciplinary Sciences: Computational Life Sciences\",\"volume\":\"15 3\",\"pages\":\"393-404\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2023-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"1\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Interdisciplinary Sciences: Computational Life Sciences\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s12539-023-00561-3\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Interdisciplinary Sciences: Computational Life Sciences","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s12539-023-00561-3","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
RNA Folding Based on 5 Beads Model and Multiscale Simulation.
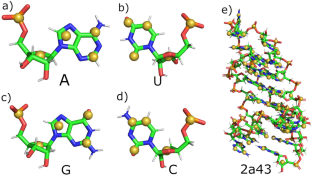
RNA folding prediction is very meaningful and challenging. The molecular dynamics simulation (MDS) of all atoms (AA) is limited to the folding of small RNA molecules. At present, most of the practical models are coarse grained (CG) model, and the coarse-grained force field (CGFF) parameters usually depend on known RNA structures. However, the limitation of the CGFF is obvious that it is difficult to study the modified RNA. Based on the 3 beads model (AIMS_RNA_B3), we proposed the AIMS_RNA_B5 model with three beads representing a base and two beads representing the main chain (sugar group and phosphate group). We first run the all atom molecular dynamic simulation (AAMDS), and fit the CGFF parameter with the AA trajectory. Then perform the coarse-grained molecular dynamic simulation (CGMDS). AAMDS is the foundation of CGMDS. CGMDS is mainly to carry out the conformation sampling based on the current AAMDS state and improve the folding speed. We simulated the folding of three RNAs, which belong to hairpin, pseudoknot and tRNA respectively. Compared to the AIMS_RNA_B3 model, the AIMS_RNA_B5 model is more reasonable and performs better.
期刊介绍:
Interdisciplinary Sciences--Computational Life Sciences aims to cover the most recent and outstanding developments in interdisciplinary areas of sciences, especially focusing on computational life sciences, an area that is enjoying rapid development at the forefront of scientific research and technology.
The journal publishes original papers of significant general interest covering recent research and developments. Articles will be published rapidly by taking full advantage of internet technology for online submission and peer-reviewing of manuscripts, and then by publishing OnlineFirstTM through SpringerLink even before the issue is built or sent to the printer.
The editorial board consists of many leading scientists with international reputation, among others, Luc Montagnier (UNESCO, France), Dennis Salahub (University of Calgary, Canada), Weitao Yang (Duke University, USA). Prof. Dongqing Wei at the Shanghai Jiatong University is appointed as the editor-in-chief; he made important contributions in bioinformatics and computational physics and is best known for his ground-breaking works on the theory of ferroelectric liquids. With the help from a team of associate editors and the editorial board, an international journal with sound reputation shall be created.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
