David S M Lee, Kathleen M Cardone, David Y Zhang, Noah L Tsao, Sarah Abramowitz, Pranav Sharma, John S DePaolo, Mitchell Conery, Krishna G Aragam, Kiran Biddinger, Ozan Dilitikas, Lily Hoffman-Andrews, Renae L Judy, Atlas Khan, Iftikhar Kulo, Megan J Puckelwartz, Nosheen Reza, Benjamin A Satterfield, Pankhuri Singhal, Zoltan P Arany, Thomas P Cappola, Eric Carruth, Sharlene M Day, Ron Do, Christopher M Haggarty, Jacob Joseph, Elizabeth M McNally, Girish Nadkarni, Anjali T Owens, Daniel J Rader, Marylyn D Ritchie, Yan V Sun, Benjamin F Voight, Michael G Levin, Scott M Damrauer
{"title":"等位基因频率谱中常见和罕见的心力衰竭变异遗传结构。","authors":"David S M Lee, Kathleen M Cardone, David Y Zhang, Noah L Tsao, Sarah Abramowitz, Pranav Sharma, John S DePaolo, Mitchell Conery, Krishna G Aragam, Kiran Biddinger, Ozan Dilitikas, Lily Hoffman-Andrews, Renae L Judy, Atlas Khan, Iftikhar Kulo, Megan J Puckelwartz, Nosheen Reza, Benjamin A Satterfield, Pankhuri Singhal, Zoltan P Arany, Thomas P Cappola, Eric Carruth, Sharlene M Day, Ron Do, Christopher M Haggarty, Jacob Joseph, Elizabeth M McNally, Girish Nadkarni, Anjali T Owens, Daniel J Rader, Marylyn D Ritchie, Yan V Sun, Benjamin F Voight, Michael G Levin, Scott M Damrauer","doi":"10.1101/2023.07.16.23292724","DOIUrl":null,"url":null,"abstract":"<p><p>Heart failure (HF) is a complex trait, influenced by environmental and genetic factors, which affects over 30 million individuals worldwide. Historically, the genetics of HF have been studied in Mendelian forms of disease, where rare genetic variants have been linked to familial cardiomyopathies. More recently, genome-wide association studies (GWAS) have successfully identified common genetic variants associated with risk of HF. However, the relative importance of genetic variants across the allele-frequency spectrum remains incompletely characterized. Here, we report the results of common- and rare-variant association studies of all-cause heart failure, applying recently developed methods to quantify the heritability of HF attributable to different classes of genetic variation. We combine GWAS data across multiple populations including 207,346 individuals with HF and 2,151,210 without, identifying 176 risk loci at genome-wide significance (P-value < 5×10<sup>-8</sup>). Signals at newly identified common-variant loci include coding variants in Mendelian cardiomyopathy genes (<i>MYBPC3</i>, <i>BAG3</i>) and in regulators of lipoprotein (<i>LPL</i>) and glucose metabolism (<i>GIPR</i>, <i>GLP1R</i>). These signals are enriched in myocyte and adipocyte cell types and can be clustered into 5 broad modules based on pleiotropic associations with anthropomorphic traits/obesity, blood pressure/renal function, atherosclerosis/lipids, immune activity, and arrhythmias. Gene burden studies across three biobanks (PMBB, UKB, AOU), including 27,208 individuals with HF and 349,126 without, uncover exome-wide significant (P-value < 1.57×10<sup>-6</sup>) associations for HF and rare predicted loss-of-function (pLoF) variants in <i>TTN</i>, <i>MYBPC3</i>, <i>FLNC, and BAG3.</i> Total burden heritability of rare coding variants (2.2%, 95% CI 0.99-3.5%) is highly concentrated in a small set of Mendelian cardiomyopathy genes, while common variant heritability (4.3%, 95% CI 3.9-4.7%) is more diffusely spread throughout the genome. Finally, we show that common-variant background, in the form of a polygenic risk score (PRS), significantly modifies the risk of HF among carriers of pathogenic truncating variants in the Mendelian cardiomyopathy gene TTN. Together, these findings provide a genetic link between dysregulated metabolism and HF, and suggest a significant polygenic component to HF exists that is not captured by current clinical genetic testing.</p>","PeriodicalId":18659,"journal":{"name":"medRxiv : the preprint server for health sciences","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-10-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/ec/53/nihpp-2023.07.16.23292724v3.PMC10371173.pdf","citationCount":"0","resultStr":"{\"title\":\"Common- and rare-variant genetic architecture of heart failure across the allele frequency spectrum.\",\"authors\":\"David S M Lee, Kathleen M Cardone, David Y Zhang, Noah L Tsao, Sarah Abramowitz, Pranav Sharma, John S DePaolo, Mitchell Conery, Krishna G Aragam, Kiran Biddinger, Ozan Dilitikas, Lily Hoffman-Andrews, Renae L Judy, Atlas Khan, Iftikhar Kulo, Megan J Puckelwartz, Nosheen Reza, Benjamin A Satterfield, Pankhuri Singhal, Zoltan P Arany, Thomas P Cappola, Eric Carruth, Sharlene M Day, Ron Do, Christopher M Haggarty, Jacob Joseph, Elizabeth M McNally, Girish Nadkarni, Anjali T Owens, Daniel J Rader, Marylyn D Ritchie, Yan V Sun, Benjamin F Voight, Michael G Levin, Scott M Damrauer\",\"doi\":\"10.1101/2023.07.16.23292724\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Heart failure (HF) is a complex trait, influenced by environmental and genetic factors, which affects over 30 million individuals worldwide. Historically, the genetics of HF have been studied in Mendelian forms of disease, where rare genetic variants have been linked to familial cardiomyopathies. More recently, genome-wide association studies (GWAS) have successfully identified common genetic variants associated with risk of HF. However, the relative importance of genetic variants across the allele-frequency spectrum remains incompletely characterized. Here, we report the results of common- and rare-variant association studies of all-cause heart failure, applying recently developed methods to quantify the heritability of HF attributable to different classes of genetic variation. We combine GWAS data across multiple populations including 207,346 individuals with HF and 2,151,210 without, identifying 176 risk loci at genome-wide significance (P-value < 5×10<sup>-8</sup>). Signals at newly identified common-variant loci include coding variants in Mendelian cardiomyopathy genes (<i>MYBPC3</i>, <i>BAG3</i>) and in regulators of lipoprotein (<i>LPL</i>) and glucose metabolism (<i>GIPR</i>, <i>GLP1R</i>). These signals are enriched in myocyte and adipocyte cell types and can be clustered into 5 broad modules based on pleiotropic associations with anthropomorphic traits/obesity, blood pressure/renal function, atherosclerosis/lipids, immune activity, and arrhythmias. Gene burden studies across three biobanks (PMBB, UKB, AOU), including 27,208 individuals with HF and 349,126 without, uncover exome-wide significant (P-value < 1.57×10<sup>-6</sup>) associations for HF and rare predicted loss-of-function (pLoF) variants in <i>TTN</i>, <i>MYBPC3</i>, <i>FLNC, and BAG3.</i> Total burden heritability of rare coding variants (2.2%, 95% CI 0.99-3.5%) is highly concentrated in a small set of Mendelian cardiomyopathy genes, while common variant heritability (4.3%, 95% CI 3.9-4.7%) is more diffusely spread throughout the genome. Finally, we show that common-variant background, in the form of a polygenic risk score (PRS), significantly modifies the risk of HF among carriers of pathogenic truncating variants in the Mendelian cardiomyopathy gene TTN. Together, these findings provide a genetic link between dysregulated metabolism and HF, and suggest a significant polygenic component to HF exists that is not captured by current clinical genetic testing.</p>\",\"PeriodicalId\":18659,\"journal\":{\"name\":\"medRxiv : the preprint server for health sciences\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2024-10-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/ec/53/nihpp-2023.07.16.23292724v3.PMC10371173.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"medRxiv : the preprint server for health sciences\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1101/2023.07.16.23292724\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"medRxiv : the preprint server for health sciences","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2023.07.16.23292724","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Common- and rare-variant genetic architecture of heart failure across the allele frequency spectrum.
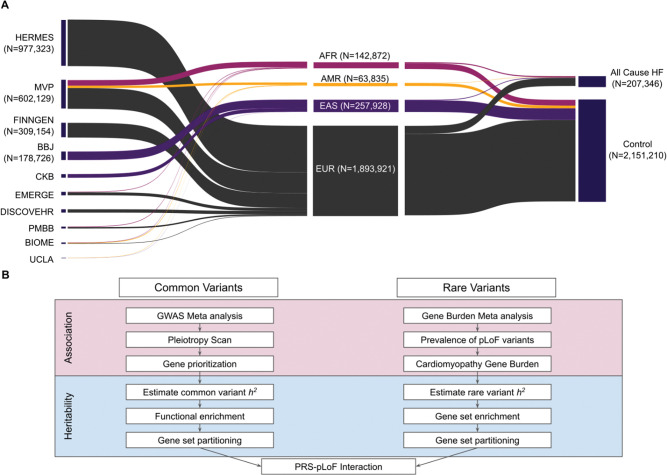
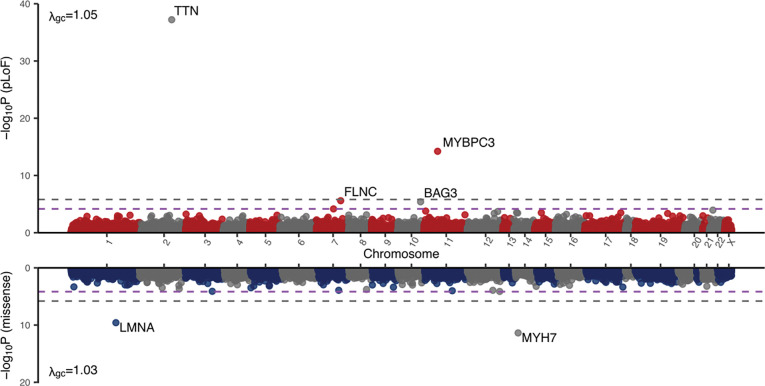
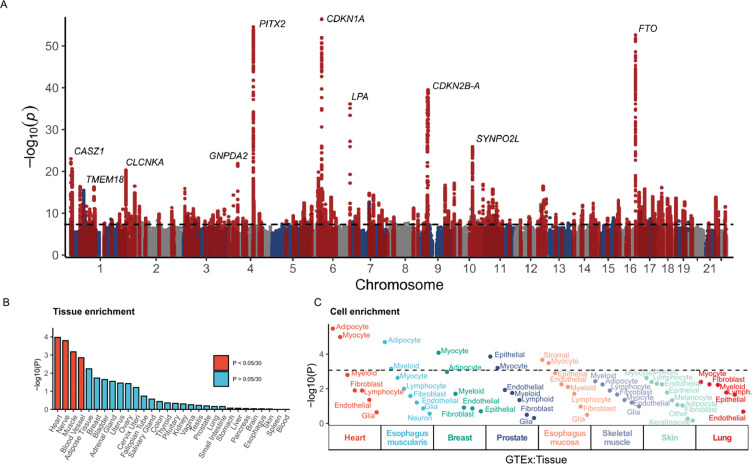
Heart failure (HF) is a complex trait, influenced by environmental and genetic factors, which affects over 30 million individuals worldwide. Historically, the genetics of HF have been studied in Mendelian forms of disease, where rare genetic variants have been linked to familial cardiomyopathies. More recently, genome-wide association studies (GWAS) have successfully identified common genetic variants associated with risk of HF. However, the relative importance of genetic variants across the allele-frequency spectrum remains incompletely characterized. Here, we report the results of common- and rare-variant association studies of all-cause heart failure, applying recently developed methods to quantify the heritability of HF attributable to different classes of genetic variation. We combine GWAS data across multiple populations including 207,346 individuals with HF and 2,151,210 without, identifying 176 risk loci at genome-wide significance (P-value < 5×10-8). Signals at newly identified common-variant loci include coding variants in Mendelian cardiomyopathy genes (MYBPC3, BAG3) and in regulators of lipoprotein (LPL) and glucose metabolism (GIPR, GLP1R). These signals are enriched in myocyte and adipocyte cell types and can be clustered into 5 broad modules based on pleiotropic associations with anthropomorphic traits/obesity, blood pressure/renal function, atherosclerosis/lipids, immune activity, and arrhythmias. Gene burden studies across three biobanks (PMBB, UKB, AOU), including 27,208 individuals with HF and 349,126 without, uncover exome-wide significant (P-value < 1.57×10-6) associations for HF and rare predicted loss-of-function (pLoF) variants in TTN, MYBPC3, FLNC, and BAG3. Total burden heritability of rare coding variants (2.2%, 95% CI 0.99-3.5%) is highly concentrated in a small set of Mendelian cardiomyopathy genes, while common variant heritability (4.3%, 95% CI 3.9-4.7%) is more diffusely spread throughout the genome. Finally, we show that common-variant background, in the form of a polygenic risk score (PRS), significantly modifies the risk of HF among carriers of pathogenic truncating variants in the Mendelian cardiomyopathy gene TTN. Together, these findings provide a genetic link between dysregulated metabolism and HF, and suggest a significant polygenic component to HF exists that is not captured by current clinical genetic testing.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
