Jan Lause, Christoph Ziegenhain, Leonard Hartmanis, Philipp Berens, Dmitry Kobak
{"title":"Compound models and Pearson residuals for single-cell RNA-seq data without UMIs.","authors":"Jan Lause, Christoph Ziegenhain, Leonard Hartmanis, Philipp Berens, Dmitry Kobak","doi":"10.1101/2023.08.02.551637","DOIUrl":null,"url":null,"abstract":"<p><p>Recent work employed Pearson residuals from Poisson or negative binomial models to normalize UMI data. To extend this approach to non-UMI data, we model the additional amplification step with a compound distribution: we assume that sequenced RNA molecules follow a negative binomial distribution, and are then replicated following an amplification distribution. We show how this model leads to compound Pearson residuals, which yield meaningful gene selection and embeddings of Smart-seq2 datasets. Further, we suggest that amplification distributions across several sequencing protocols can be described by a broken power law. The resulting compound model captures previously unexplained overdispersion and zero-inflation patterns in non-UMI data.</p>","PeriodicalId":72407,"journal":{"name":"bioRxiv : the preprint server for biology","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-07-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://ftp.ncbi.nlm.nih.gov/pub/pmc/oa_pdf/16/c4/nihpp-2023.08.02.551637v1.PMC10418209.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"bioRxiv : the preprint server for biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2023.08.02.551637","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
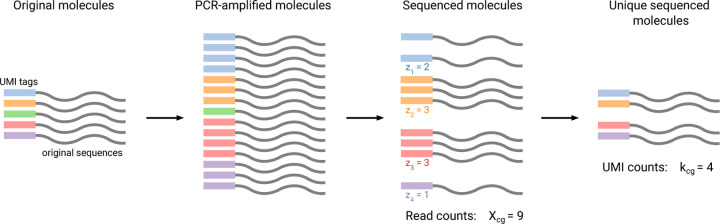
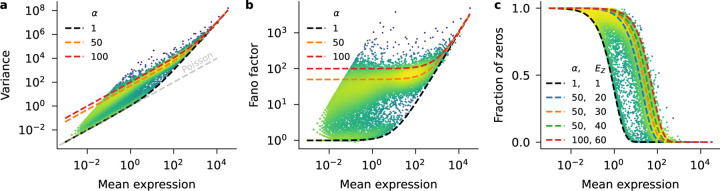
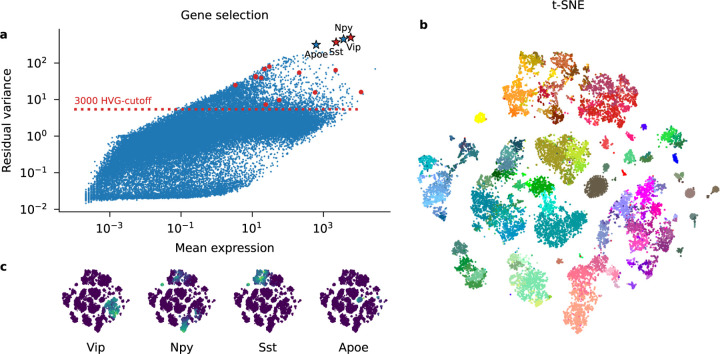
Recent work employed Pearson residuals from Poisson or negative binomial models to normalize UMI data. To extend this approach to non-UMI data, we model the additional amplification step with a compound distribution: we assume that sequenced RNA molecules follow a negative binomial distribution, and are then replicated following an amplification distribution. We show how this model leads to compound Pearson residuals, which yield meaningful gene selection and embeddings of Smart-seq2 datasets. Further, we suggest that amplification distributions across several sequencing protocols can be described by a broken power law. The resulting compound model captures previously unexplained overdispersion and zero-inflation patterns in non-UMI data.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
