Caveolins are the principal structural components of plasma membrane caveolae. Dominant pathogenic mutations in the muscle-specific caveolin-3 (Cav3) gene isoform, such as the limb girdle muscular dystrophy type 1C (LGMD-1C) P104L mutation, result in dramatic loss of the Cav3 protein and pathophysiological muscle weakness/wasting. We hypothesize that such muscle degeneration may be linked to disturbances in signalling events that impact protein turnover. Herein, we report studies assessing the effects of Cav3 deficiency on mammalian or mechanistic target of rapamycin complex 1 (mTORC1) signalling in skeletal muscle cells.
L6 myoblasts were stably transfected with Cav3P104L or expression of native Cav3 was abolished by CRISPR/Cas9 genome editing (Cav3 knockout [Cav3KO]) prior to performing subcellular fractionation and immunoblotting, analysis of real-time mitochondrial respiration or fixed cell immunocytochemistry. Skeletal muscle from wild-type and Cav3−/− mice was processed for immunoblot analysis of downstream mTORC1 substrate phosphorylation.
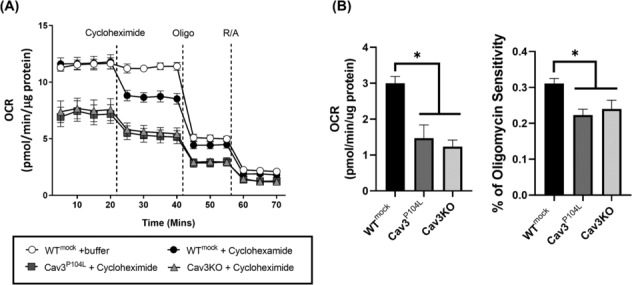
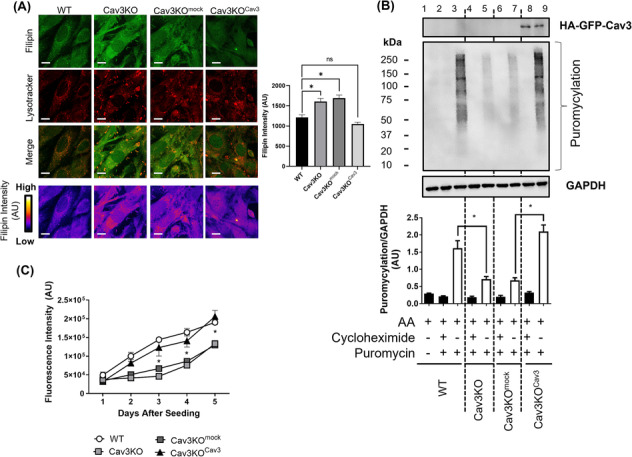
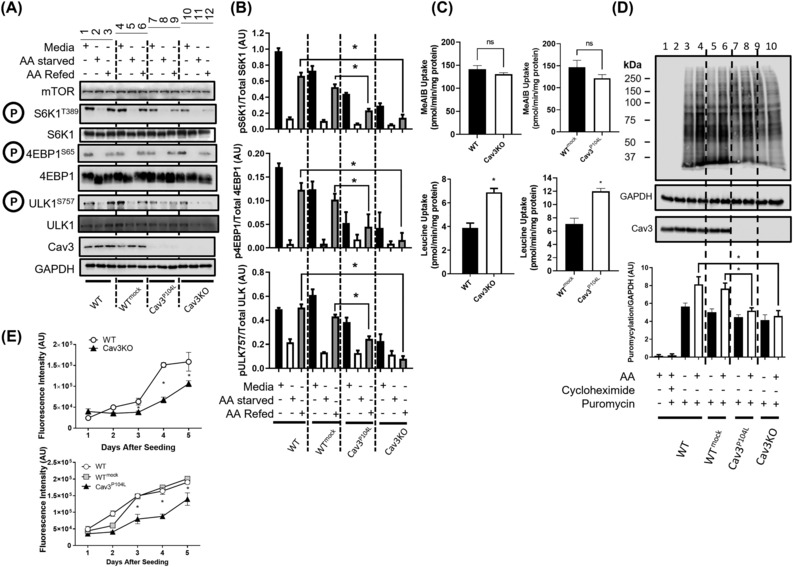
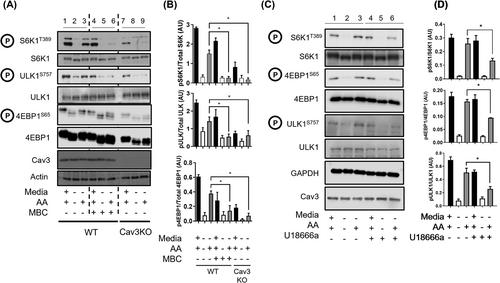
Cav3 was detected in lysosomal-enriched membranes isolated from L6 myoblasts and observed by confocal microscopy to co-localize with lysosomal-specific markers. Cav3P104L expression, which results in significant (~95%) loss of native Cav3, or CRISPR/Cas9-mediated Cav3KO, reduced amino acid-dependent mTORC1 activation. The decline in mTORC1-directed signalling was detected by immunoblot analysis of L6 muscle cells and gastrocnemius Cav3−/− mouse muscle as judged by reduced phosphorylation of mTORC1 substrates that play key roles in the initiation of protein synthesis (4EBP1S65 and S6K1T389). S6K1T389 and 4EBP1S65 phosphorylation reduced by over 75% and 80% in Cav3KO muscle cells and by over 90% and 30% in Cav3−/− mouse skeletal muscle, respectively. The reduction in protein synthetic capacity in L6 muscle cells was confirmed by analysis of puromycylated peptides using the SUnSET assay. Cav3 loss was also associated with a 26% increase in lysosomal cholesterol, and pharmacological manipulation of lysosomal cholesterol was effective in replicating the reduction in mTORC1 activity observed in Cav3KO cells. Notably, re-expression of Cav3 in Cav3KO myoblasts normalized lysosomal cholesterol content, which coincided with a recovery in protein translation and an associated increase in mTORC1-directed phosphorylation of downstream targets.
Our findings indicate that Cav3 can localize on lysosomal membranes and is a novel regulator of mTORC1 signalling in muscle. Cav3 deficiency associated with the Cav3P104L mutation impairs mTORC1 activation and protein synthetic capacity in skeletal muscle cells, which may be linked to disturbances in lysosomal cholesterol trafficking and contribute to the pathology of LGMD-1C.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
