{"title":"Evaluating the Accuracy of Methods for Detecting Correlated Rates of Molecular and Morphological Evolution.","authors":"Yasmin Asar, Hervé Sauquet, Simon Y W Ho","doi":"10.1093/sysbio/syad055","DOIUrl":null,"url":null,"abstract":"<p><p>Determining the link between genomic and phenotypic change is a fundamental goal in evolutionary biology. Insights into this link can be gained by using a phylogenetic approach to test for correlations between rates of molecular and morphological evolution. However, there has been persistent uncertainty about the relationship between these rates, partly because conflicting results have been obtained using various methods that have not been examined in detail. We carried out a simulation study to evaluate the performance of 5 statistical methods for detecting correlated rates of evolution. Our simulations explored the evolution of molecular sequences and morphological characters under a range of conditions. Of the methods tested, Bayesian relaxed-clock estimation of branch rates was able to detect correlated rates of evolution correctly in the largest number of cases. This was followed by correlations of root-to-tip distances, Bayesian model selection, independent sister-pairs contrasts, and likelihood-based model selection. As expected, the power to detect correlated rates increased with the amount of data, both in terms of tree size and number of morphological characters. Likewise, greater among-lineage rate variation in the data led to improved performance of all 5 methods, particularly for Bayesian relaxed-clock analysis when the rate model was mismatched. We then applied these methods to a data set from flowering plants and did not find evidence of a correlation in evolutionary rates between genomic data and morphological characters. The results of our study have practical implications for phylogenetic analyses of combined molecular and morphological data sets, and highlight the conditions under which the links between genomic and phenotypic rates of evolution can be evaluated quantitatively.</p>","PeriodicalId":22120,"journal":{"name":"Systematic Biology","volume":" ","pages":"1337-1356"},"PeriodicalIF":5.7000,"publicationDate":"2023-12-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10924723/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Systematic Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/sysbio/syad055","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"EVOLUTIONARY BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
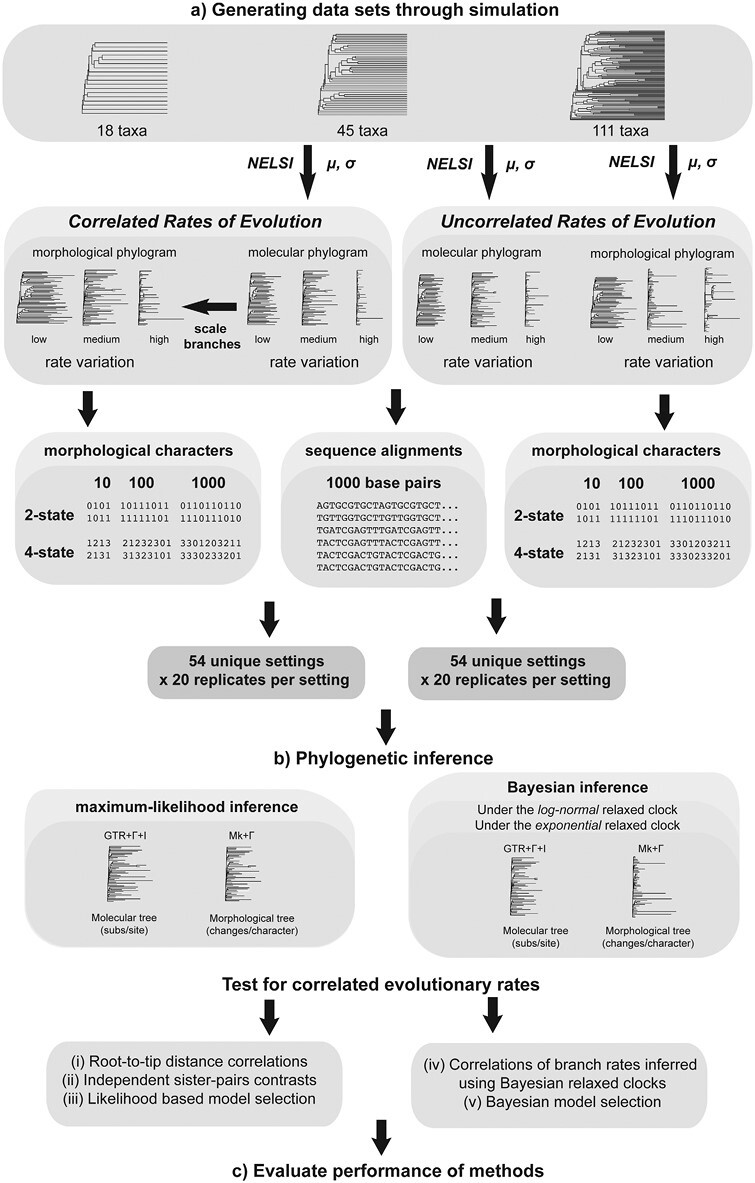
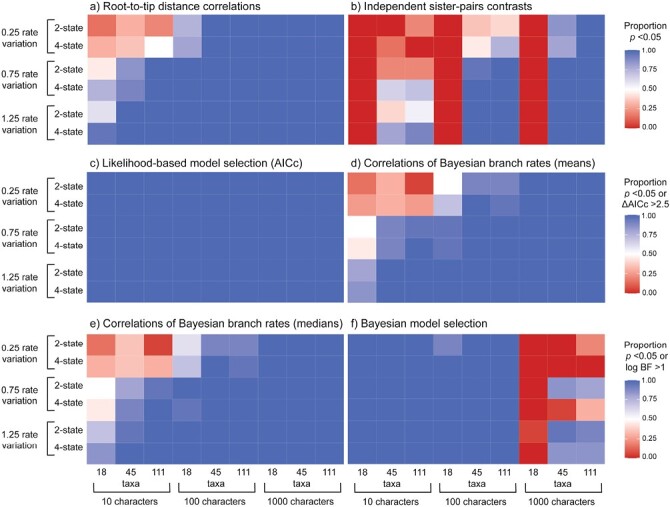
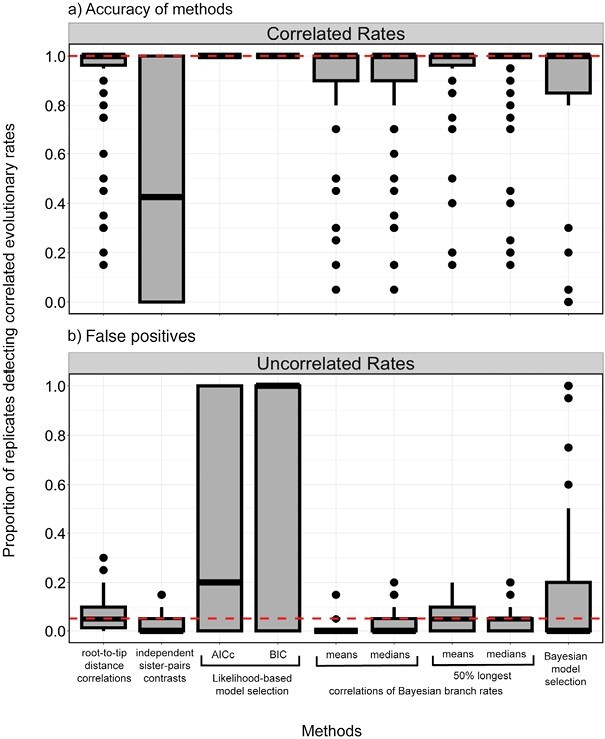
Determining the link between genomic and phenotypic change is a fundamental goal in evolutionary biology. Insights into this link can be gained by using a phylogenetic approach to test for correlations between rates of molecular and morphological evolution. However, there has been persistent uncertainty about the relationship between these rates, partly because conflicting results have been obtained using various methods that have not been examined in detail. We carried out a simulation study to evaluate the performance of 5 statistical methods for detecting correlated rates of evolution. Our simulations explored the evolution of molecular sequences and morphological characters under a range of conditions. Of the methods tested, Bayesian relaxed-clock estimation of branch rates was able to detect correlated rates of evolution correctly in the largest number of cases. This was followed by correlations of root-to-tip distances, Bayesian model selection, independent sister-pairs contrasts, and likelihood-based model selection. As expected, the power to detect correlated rates increased with the amount of data, both in terms of tree size and number of morphological characters. Likewise, greater among-lineage rate variation in the data led to improved performance of all 5 methods, particularly for Bayesian relaxed-clock analysis when the rate model was mismatched. We then applied these methods to a data set from flowering plants and did not find evidence of a correlation in evolutionary rates between genomic data and morphological characters. The results of our study have practical implications for phylogenetic analyses of combined molecular and morphological data sets, and highlight the conditions under which the links between genomic and phenotypic rates of evolution can be evaluated quantitatively.
期刊介绍:
Systematic Biology is the bimonthly journal of the Society of Systematic Biologists. Papers for the journal are original contributions to the theory, principles, and methods of systematics as well as phylogeny, evolution, morphology, biogeography, paleontology, genetics, and the classification of all living things. A Points of View section offers a forum for discussion, while book reviews and announcements of general interest are also featured.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
