Sarah N. Elliott, Kevin B. Moore III, Clayton R. Mulvihill, Andreas V. Copan, Luna Pratali Maffei, Stephen J. Klippenstein
{"title":"The role of stereochemistry in combustion processes","authors":"Sarah N. Elliott, Kevin B. Moore III, Clayton R. Mulvihill, Andreas V. Copan, Luna Pratali Maffei, Stephen J. Klippenstein","doi":"10.1002/wcms.1710","DOIUrl":null,"url":null,"abstract":"<p>Stereochemical effects significantly influence chemical processes, yet it is not well understood if they are a leading source of uncertainty in combustion modeling. Stereochemistry influences a combustion model (i) at the earliest stage of its construction when mapping the reaction network, (ii) in the computation of individual thermochemical and rate parameters, and (iii) in the prediction of combustion observables. The present work reviews the importance of enumerating stereochemical species and reactions at each of these steps. Further, it analyzes the separate influence of several types of stereochemistry, including geometric, optical, and fleeting transition state diastereomers. Three reaction networks serve to examine which stages of low-temperature oxidation are most affected by stereochemistry, including the first and second oxidation of <i>n</i>-butane, the third oxidation of <i>n</i>-pentane, and the early stages of pyrolysis of 1- and 2-pentene. The 149 reactions in the <i>n</i>-butane mechanism are expanded to 183 reactions when accounting for diastereomerism. Each of these 183 reactions is parameterized with ab initio kinetics computations to determine that, for the <i>n</i>-butane mechanism, the median factor of diastereomeric deviation is 3.5 at 360 K for rate constants and as high as 1.6 for mechanism reactivity, in terms of ignition delay times, as opposed to a mechanism without stereochemical expansion.</p><p>This article is categorized under:\n </p>","PeriodicalId":236,"journal":{"name":"Wiley Interdisciplinary Reviews: Computational Molecular Science","volume":"14 2","pages":""},"PeriodicalIF":10.9000,"publicationDate":"2024-03-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/wcms.1710","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Wiley Interdisciplinary Reviews: Computational Molecular Science","FirstCategoryId":"92","ListUrlMain":"https://wires.onlinelibrary.wiley.com/doi/10.1002/wcms.1710","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
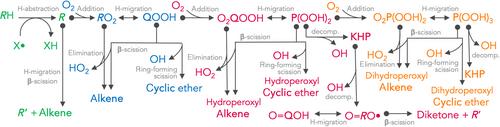
Stereochemical effects significantly influence chemical processes, yet it is not well understood if they are a leading source of uncertainty in combustion modeling. Stereochemistry influences a combustion model (i) at the earliest stage of its construction when mapping the reaction network, (ii) in the computation of individual thermochemical and rate parameters, and (iii) in the prediction of combustion observables. The present work reviews the importance of enumerating stereochemical species and reactions at each of these steps. Further, it analyzes the separate influence of several types of stereochemistry, including geometric, optical, and fleeting transition state diastereomers. Three reaction networks serve to examine which stages of low-temperature oxidation are most affected by stereochemistry, including the first and second oxidation of n-butane, the third oxidation of n-pentane, and the early stages of pyrolysis of 1- and 2-pentene. The 149 reactions in the n-butane mechanism are expanded to 183 reactions when accounting for diastereomerism. Each of these 183 reactions is parameterized with ab initio kinetics computations to determine that, for the n-butane mechanism, the median factor of diastereomeric deviation is 3.5 at 360 K for rate constants and as high as 1.6 for mechanism reactivity, in terms of ignition delay times, as opposed to a mechanism without stereochemical expansion.
期刊介绍:
Computational molecular sciences harness the power of rigorous chemical and physical theories, employing computer-based modeling, specialized hardware, software development, algorithm design, and database management to explore and illuminate every facet of molecular sciences. These interdisciplinary approaches form a bridge between chemistry, biology, and materials sciences, establishing connections with adjacent application-driven fields in both chemistry and biology. WIREs Computational Molecular Science stands as a platform to comprehensively review and spotlight research from these dynamic and interconnected fields.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
