Edgar Josué Landinez Borda, Kenneth O. Berard, Annette Lopez and Brenda Rubenstein
{"title":"Gaussian processes for finite size extrapolation of many-body simulations†","authors":"Edgar Josué Landinez Borda, Kenneth O. Berard, Annette Lopez and Brenda Rubenstein","doi":"10.1039/D4FD00051J","DOIUrl":null,"url":null,"abstract":"<p >Key to being able to accurately model the properties of realistic materials is being able to predict their properties in the thermodynamic limit. Nevertheless, because most many-body electronic structure methods scale as a high-order polynomial, or even exponentially, with system size, directly simulating large systems in their thermodynamic limit rapidly becomes computationally intractable. As a result, researchers typically estimate the properties of large systems that approach the thermodynamic limit by extrapolating the properties of smaller, computationally-accessible systems based on relatively simple scaling expressions. In this work, we employ Gaussian processes to more accurately and efficiently extrapolate many-body simulations to their thermodynamic limit. We train our Gaussian processes on Smooth Overlap of Atomic Positions (SOAP) descriptors to extrapolate the energies of one-dimensional hydrogen chains obtained using two high-accuracy many-body methods: coupled cluster theory and Auxiliary Field Quantum Monte Carlo (AFQMC). In so doing, we show that Gaussian processes trained on relatively short 10–30-atom chains can predict the energies of both homogeneous and inhomogeneous hydrogen chains in their thermodynamic limit with sub-milliHartree accuracy. Unlike standard scaling expressions, our GPR-based approach is highly generalizable given representative training data and is not dependent on systems’ geometries or dimensionality. This work highlights the potential for machine learning to correct for the finite size effects that routinely complicate the interpretation of finite size many-body simulations.</p>","PeriodicalId":49075,"journal":{"name":"Faraday Discussions","volume":"254 ","pages":" 500-528"},"PeriodicalIF":3.1000,"publicationDate":"2024-03-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/fd/d4fd00051j?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Faraday Discussions","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/fd/d4fd00051j","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Chemistry","Score":null,"Total":0}
引用次数: 0
Abstract
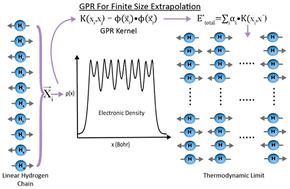
Key to being able to accurately model the properties of realistic materials is being able to predict their properties in the thermodynamic limit. Nevertheless, because most many-body electronic structure methods scale as a high-order polynomial, or even exponentially, with system size, directly simulating large systems in their thermodynamic limit rapidly becomes computationally intractable. As a result, researchers typically estimate the properties of large systems that approach the thermodynamic limit by extrapolating the properties of smaller, computationally-accessible systems based on relatively simple scaling expressions. In this work, we employ Gaussian processes to more accurately and efficiently extrapolate many-body simulations to their thermodynamic limit. We train our Gaussian processes on Smooth Overlap of Atomic Positions (SOAP) descriptors to extrapolate the energies of one-dimensional hydrogen chains obtained using two high-accuracy many-body methods: coupled cluster theory and Auxiliary Field Quantum Monte Carlo (AFQMC). In so doing, we show that Gaussian processes trained on relatively short 10–30-atom chains can predict the energies of both homogeneous and inhomogeneous hydrogen chains in their thermodynamic limit with sub-milliHartree accuracy. Unlike standard scaling expressions, our GPR-based approach is highly generalizable given representative training data and is not dependent on systems’ geometries or dimensionality. This work highlights the potential for machine learning to correct for the finite size effects that routinely complicate the interpretation of finite size many-body simulations.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
