{"title":"Investigating the catalytic activity of Mgn (n = 4–8) clusters for the hydrogen evolution reaction using density functional theory","authors":"Jing Jiang, Shunping Shi, Xiaofeng Zhao, Zhanjiang Duan, Jiabao Hu, Leilei Tang, Ruixiao Yang, Jing Yang","doi":"10.1002/qua.27383","DOIUrl":null,"url":null,"abstract":"<p>To efficiently desorb H<sub>2</sub>, pure Mg<sub><i>n</i></sub> (<i>n</i> = 4–8) clusters were chosen for the hydrogen evolution reaction with H<sub>2</sub>O. At the PBE0/def2-TZVP level and the PBE0-D3/def2-TZVP level, the lowest energy structures of Mg<sub><i>n</i></sub> (<i>n</i> = 4–8) clusters and the most stable structures of Mg<sub><i>n</i></sub>@H<sub>2</sub>O (<i>n</i> = 4–8) complexes were searched in the local region. The transition state was predicted, and then the hydrogen evolution reaction channel was obtained by using the intrinsic reaction coordinate (IRC) to confirm the transition state. To better analyze the hydrogen reaction mechanism, the character of Mg<sub><i>n</i></sub>@H<sub>2</sub>O (<i>n</i> = 4–8) complexes and Mg<sub><i>n</i></sub>O (<i>n</i> = 4–8) clusters, as well as the atomic charge change trend, were investigated using interaction region indicator function analysis (IRI) and natural population analysis (NPA). The reaction effect of Mg<sub>4</sub> cluster and H<sub>2</sub>O is the worst. The energy barrier does, however, progressively lower as the cluster atom count rises, improving the reaction effect.</p>","PeriodicalId":182,"journal":{"name":"International Journal of Quantum Chemistry","volume":"124 9","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2024-04-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Quantum Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/qua.27383","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
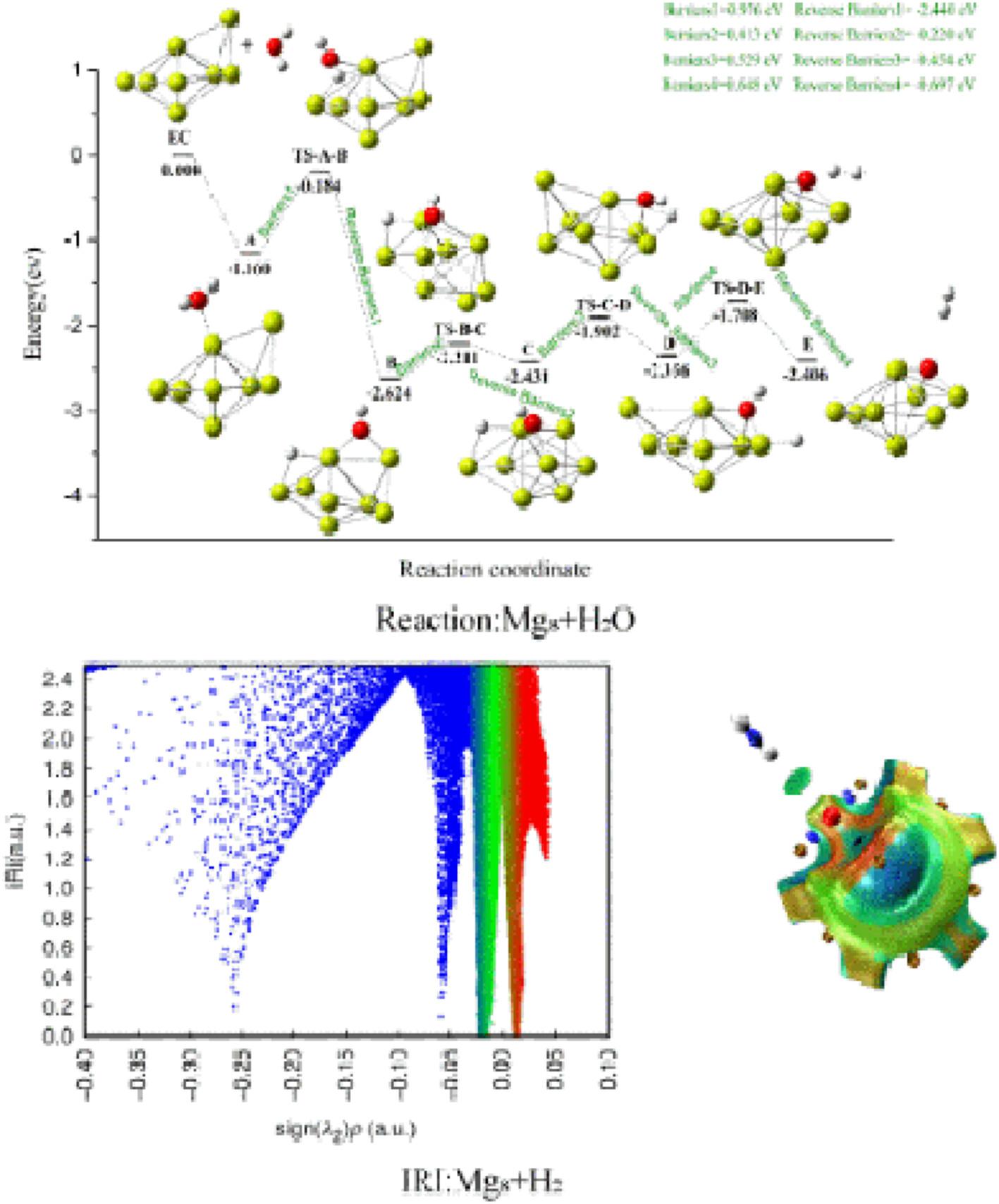
To efficiently desorb H2, pure Mgn (n = 4–8) clusters were chosen for the hydrogen evolution reaction with H2O. At the PBE0/def2-TZVP level and the PBE0-D3/def2-TZVP level, the lowest energy structures of Mgn (n = 4–8) clusters and the most stable structures of Mgn@H2O (n = 4–8) complexes were searched in the local region. The transition state was predicted, and then the hydrogen evolution reaction channel was obtained by using the intrinsic reaction coordinate (IRC) to confirm the transition state. To better analyze the hydrogen reaction mechanism, the character of Mgn@H2O (n = 4–8) complexes and MgnO (n = 4–8) clusters, as well as the atomic charge change trend, were investigated using interaction region indicator function analysis (IRI) and natural population analysis (NPA). The reaction effect of Mg4 cluster and H2O is the worst. The energy barrier does, however, progressively lower as the cluster atom count rises, improving the reaction effect.
期刊介绍:
Since its first formulation quantum chemistry has provided the conceptual and terminological framework necessary to understand atoms, molecules and the condensed matter. Over the past decades synergistic advances in the methodological developments, software and hardware have transformed quantum chemistry in a truly interdisciplinary science that has expanded beyond its traditional core of molecular sciences to fields as diverse as chemistry and catalysis, biophysics, nanotechnology and material science.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
