The Conformation of Glycosidic Linkages According to Various Force Fields: Monte Carlo Modeling of Polysaccharides Based on Extrapolation of Short-Chain Properties.
{"title":"The Conformation of Glycosidic Linkages According to Various Force Fields: Monte Carlo Modeling of Polysaccharides Based on Extrapolation of Short-Chain Properties.","authors":"Valery Lutsyk, Pawel Wolski, Wojciech Plazinski","doi":"10.1021/acs.jctc.4c00543","DOIUrl":null,"url":null,"abstract":"<p><p>The conformational features of the glycosidic linkage are the most important variable to consider when studying di-, oligo-, and polysaccharide molecules using molecular dynamics (MD) simulations. The accuracy of the theoretical model describing this degree of freedom influences the quality of the results obtained from MD calculations based on this model. This article focuses on the following two issues related to the conformation of the glycosidic linkage. First, we describe the results of a comparative analysis of the predictions of three carbohydrate-dedicated classical force fields for MD simulations, namely, CHARMM, GLYCAM, and GROMOS, in the context of different parameters of structural and energetic nature related to the conformation of selected types of glycosidic linkages, α(1 → 4), β(1 → 3), and β(1 → 4), connecting glucopyranose units. This analysis revealed several differences, mainly concerning the energy levels of the secondary and tertiary conformers and the linkage flexibility within the dominant <i>exo</i>-<i>syn</i> conformation for α(1 → 4) and β(1 → 3) linkages. Some aspects of the comparative analysis also included the newly developed, carbohydrate-dedicated Martini 3 coarse-grained force field. Second, to overcome the time-scale problem associated with sampling slow degrees of freedom in polysaccharide chains during MD simulations, we developed a coarse-grained (CG) model based on the data from MD simulations and designed for Monte Carlo modeling. This model (CG MC) is based on information from simulations of short saccharide chains, effectively sampled in atomistic MD simulations, and is capable of extrapolating local conformational properties to the case of polysaccharides of arbitrary length. The CG MC model has the potential to estimate the conformations of very long polysaccharide chains, taking into account the influence of secondary and tertiary conformations of glycosidic linkages. With respect to the comparative analysis of force fields, the application of CG MC modeling showed that relatively small differences in the predictions of individual force fields with respect to a single glycosidic linkage accumulate when considering their effect on the structure of longer chains, leading to drastically different predictions with respect to parameters describing the polymer conformation, such as the persistence length.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"6350-6368"},"PeriodicalIF":5.5000,"publicationDate":"2024-07-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11270825/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00543","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/7/10 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
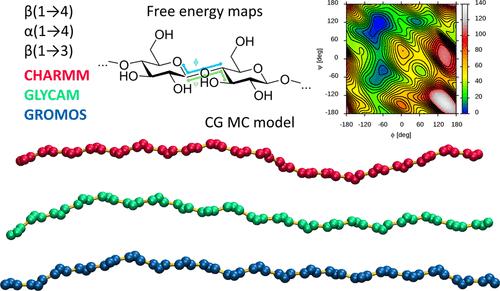
The conformational features of the glycosidic linkage are the most important variable to consider when studying di-, oligo-, and polysaccharide molecules using molecular dynamics (MD) simulations. The accuracy of the theoretical model describing this degree of freedom influences the quality of the results obtained from MD calculations based on this model. This article focuses on the following two issues related to the conformation of the glycosidic linkage. First, we describe the results of a comparative analysis of the predictions of three carbohydrate-dedicated classical force fields for MD simulations, namely, CHARMM, GLYCAM, and GROMOS, in the context of different parameters of structural and energetic nature related to the conformation of selected types of glycosidic linkages, α(1 → 4), β(1 → 3), and β(1 → 4), connecting glucopyranose units. This analysis revealed several differences, mainly concerning the energy levels of the secondary and tertiary conformers and the linkage flexibility within the dominant exo-syn conformation for α(1 → 4) and β(1 → 3) linkages. Some aspects of the comparative analysis also included the newly developed, carbohydrate-dedicated Martini 3 coarse-grained force field. Second, to overcome the time-scale problem associated with sampling slow degrees of freedom in polysaccharide chains during MD simulations, we developed a coarse-grained (CG) model based on the data from MD simulations and designed for Monte Carlo modeling. This model (CG MC) is based on information from simulations of short saccharide chains, effectively sampled in atomistic MD simulations, and is capable of extrapolating local conformational properties to the case of polysaccharides of arbitrary length. The CG MC model has the potential to estimate the conformations of very long polysaccharide chains, taking into account the influence of secondary and tertiary conformations of glycosidic linkages. With respect to the comparative analysis of force fields, the application of CG MC modeling showed that relatively small differences in the predictions of individual force fields with respect to a single glycosidic linkage accumulate when considering their effect on the structure of longer chains, leading to drastically different predictions with respect to parameters describing the polymer conformation, such as the persistence length.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
