{"title":"Newborn screening for acid sphingomyelinase deficiency in Illinois: A single center's experience","authors":"Rachel E. Hickey, Joshua Baker","doi":"10.1002/jimd.12780","DOIUrl":null,"url":null,"abstract":"<p>Acid sphingomyelinase deficiency (ASMD) is a rare lysosomal storage disorder (LSD) caused by reduced activity of the acid sphingomyelinase (ASM) enzyme, which leads to progressive storage of sphingomyelin and related lipids in the body. ASMD is caused by biallelic variants in the <i>SMPD1</i> gene, which encodes for the ASM enzyme. Current estimates of disease incidence range from 0.4 to 0.6 in 100 000 livebirths, although this is likely an underestimation of the true frequency of the disorder. While there is no cure for ASMD, comprehensive care guidelines and enzyme replacement therapy are available, making an early diagnosis crucial. Newborn screening (NBS) for ASMD is possible through measurement of ASM activity in dried blood spots and offers the opportunity for early diagnosis. In 2015, Illinois (IL) became the first to initiate statewide implementation of NBS for ASMD. This study describes the outcomes of screen-positive patients referred to Ann & Robert H. Lurie Children's Hospital (Lurie). Ten infants were referred for diagnostic evaluation at Lurie, and all 10 infants were classified as confirmed ASMD or at risk for ASMD through a combination of molecular and biochemical testing. Disease incidence was calculated using data from this statewide implementation program and was ~0.79 in 100 000 livebirths. This study demonstrates successful implementation of NBS for ASMD in IL, with high screen specificity and a notable absence of false positive screens.</p>","PeriodicalId":16281,"journal":{"name":"Journal of Inherited Metabolic Disease","volume":"47 6","pages":"1363-1370"},"PeriodicalIF":3.8000,"publicationDate":"2024-07-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jimd.12780","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Inherited Metabolic Disease","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jimd.12780","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
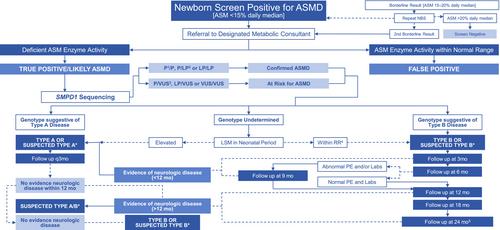
Acid sphingomyelinase deficiency (ASMD) is a rare lysosomal storage disorder (LSD) caused by reduced activity of the acid sphingomyelinase (ASM) enzyme, which leads to progressive storage of sphingomyelin and related lipids in the body. ASMD is caused by biallelic variants in the SMPD1 gene, which encodes for the ASM enzyme. Current estimates of disease incidence range from 0.4 to 0.6 in 100 000 livebirths, although this is likely an underestimation of the true frequency of the disorder. While there is no cure for ASMD, comprehensive care guidelines and enzyme replacement therapy are available, making an early diagnosis crucial. Newborn screening (NBS) for ASMD is possible through measurement of ASM activity in dried blood spots and offers the opportunity for early diagnosis. In 2015, Illinois (IL) became the first to initiate statewide implementation of NBS for ASMD. This study describes the outcomes of screen-positive patients referred to Ann & Robert H. Lurie Children's Hospital (Lurie). Ten infants were referred for diagnostic evaluation at Lurie, and all 10 infants were classified as confirmed ASMD or at risk for ASMD through a combination of molecular and biochemical testing. Disease incidence was calculated using data from this statewide implementation program and was ~0.79 in 100 000 livebirths. This study demonstrates successful implementation of NBS for ASMD in IL, with high screen specificity and a notable absence of false positive screens.
期刊介绍:
The Journal of Inherited Metabolic Disease (JIMD) is the official journal of the Society for the Study of Inborn Errors of Metabolism (SSIEM). By enhancing communication between workers in the field throughout the world, the JIMD aims to improve the management and understanding of inherited metabolic disorders. It publishes results of original research and new or important observations pertaining to any aspect of inherited metabolic disease in humans and higher animals. This includes clinical (medical, dental and veterinary), biochemical, genetic (including cytogenetic, molecular and population genetic), experimental (including cell biological), methodological, theoretical, epidemiological, ethical and counselling aspects. The JIMD also reviews important new developments or controversial issues relating to metabolic disorders and publishes reviews and short reports arising from the Society''s annual symposia. A distinction is made between peer-reviewed scientific material that is selected because of its significance for other professionals in the field and non-peer- reviewed material that aims to be important, controversial, interesting or entertaining (“Extras”).



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
