{"title":"Automated fiducial-based alignment of cryo-electron tomography tilt series in Dynamo","authors":"","doi":"10.1016/j.str.2024.07.003","DOIUrl":null,"url":null,"abstract":"<p>With the advent of modern technologies for cryo-electron tomography (cryo-ET), high-quality tilt series are more rapidly acquired than processed and analyzed. Thus, a robust and fast-automated alignment for batch processing in cryo-ET is needed. While different software packages have made available several approaches for automated marker-based alignment of tilt series, manual user intervention remains necessary for many datasets, thus preventing high-throughput tomography.</p><p>We have developed a MATLAB-based framework integrated into the Dynamo software package for automatic detection of fiducial markers that generates a robust alignment model with minimal input parameters. This approach allows high-throughput, unsupervised volume reconstruction. This new module extends Dynamo with a large repertory of tools for tomographic alignment and reconstruction, as well as specific visualization browsers to rapidly assess the biological relevance of the dataset. Our approach has been successfully tested on a broad range of datasets that include diverse biological samples and cryo-ET modalities.</p>","PeriodicalId":22168,"journal":{"name":"Structure","volume":"19 1","pages":""},"PeriodicalIF":4.3000,"publicationDate":"2024-07-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Structure","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.str.2024.07.003","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
With the advent of modern technologies for cryo-electron tomography (cryo-ET), high-quality tilt series are more rapidly acquired than processed and analyzed. Thus, a robust and fast-automated alignment for batch processing in cryo-ET is needed. While different software packages have made available several approaches for automated marker-based alignment of tilt series, manual user intervention remains necessary for many datasets, thus preventing high-throughput tomography.
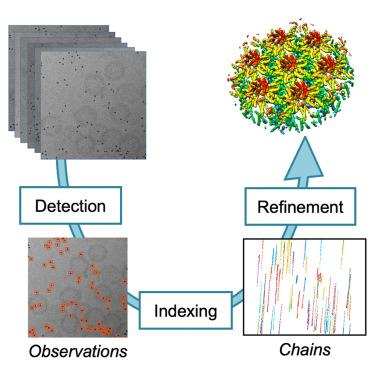
We have developed a MATLAB-based framework integrated into the Dynamo software package for automatic detection of fiducial markers that generates a robust alignment model with minimal input parameters. This approach allows high-throughput, unsupervised volume reconstruction. This new module extends Dynamo with a large repertory of tools for tomographic alignment and reconstruction, as well as specific visualization browsers to rapidly assess the biological relevance of the dataset. Our approach has been successfully tested on a broad range of datasets that include diverse biological samples and cryo-ET modalities.
期刊介绍:
Structure aims to publish papers of exceptional interest in the field of structural biology. The journal strives to be essential reading for structural biologists, as well as biologists and biochemists that are interested in macromolecular structure and function. Structure strongly encourages the submission of manuscripts that present structural and molecular insights into biological function and mechanism. Other reports that address fundamental questions in structural biology, such as structure-based examinations of protein evolution, folding, and/or design, will also be considered. We will consider the application of any method, experimental or computational, at high or low resolution, to conduct structural investigations, as long as the method is appropriate for the biological, functional, and mechanistic question(s) being addressed. Likewise, reports describing single-molecule analysis of biological mechanisms are welcome.
In general, the editors encourage submission of experimental structural studies that are enriched by an analysis of structure-activity relationships and will not consider studies that solely report structural information unless the structure or analysis is of exceptional and broad interest. Studies reporting only homology models, de novo models, or molecular dynamics simulations are also discouraged unless the models are informed by or validated by novel experimental data; rationalization of a large body of existing experimental evidence and making testable predictions based on a model or simulation is often not considered sufficient.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
