Sean J. Jurgens, Xin Wang, Seung Hoan Choi, Lu-Chen Weng, Satoshi Koyama, James P. Pirruccello, Trang Nguyen, Patrick Smadbeck, Dongkeun Jang, Mark Chaffin, Roddy Walsh, Carolina Roselli, Amanda L. Elliott, Leonoor F. J. M. Wijdeveld, Kiran J. Biddinger, Shinwan Kany, Joel T. Rämö, Pradeep Natarajan, Krishna G. Aragam, Jason Flannick, Noël P. Burtt, Connie R. Bezzina, Steven A. Lubitz, Kathryn L. Lunetta, Patrick T. Ellinor
{"title":"Rare coding variant analysis for human diseases across biobanks and ancestries","authors":"Sean J. Jurgens, Xin Wang, Seung Hoan Choi, Lu-Chen Weng, Satoshi Koyama, James P. Pirruccello, Trang Nguyen, Patrick Smadbeck, Dongkeun Jang, Mark Chaffin, Roddy Walsh, Carolina Roselli, Amanda L. Elliott, Leonoor F. J. M. Wijdeveld, Kiran J. Biddinger, Shinwan Kany, Joel T. Rämö, Pradeep Natarajan, Krishna G. Aragam, Jason Flannick, Noël P. Burtt, Connie R. Bezzina, Steven A. Lubitz, Kathryn L. Lunetta, Patrick T. Ellinor","doi":"10.1038/s41588-024-01894-5","DOIUrl":null,"url":null,"abstract":"Large-scale sequencing has enabled unparalleled opportunities to investigate the role of rare coding variation in human phenotypic variability. Here, we present a pan-ancestry analysis of sequencing data from three large biobanks, including the All of Us research program. Using mixed-effects models, we performed gene-based rare variant testing for 601 diseases across 748,879 individuals, including 155,236 with ancestry dissimilar to European. We identified 363 significant associations, which highlighted core genes for the human disease phenome and identified potential novel associations, including UBR3 for cardiometabolic disease and YLPM1 for psychiatric disease. Pan-ancestry burden testing represented an inclusive and useful approach for discovery in diverse datasets, although we also highlight the importance of ancestry-specific sensitivity analyses in this setting. Finally, we found that effect sizes for rare protein-disrupting variants were concordant between samples similar to European ancestry and other genetic ancestries (βDeming = 0.7–1.0). Our results have implications for multi-ancestry and cross-biobank approaches in sequencing association studies for human disease. Gene-based rare variant analyses for 601 diseases across 748,879 individuals from three biobanks identify 363 significant associations and highlight important considerations for multi-ancestry and cross-biobank sequencing studies.","PeriodicalId":18985,"journal":{"name":"Nature genetics","volume":"56 9","pages":"1811-1820"},"PeriodicalIF":29.0000,"publicationDate":"2024-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41588-024-01894-5","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
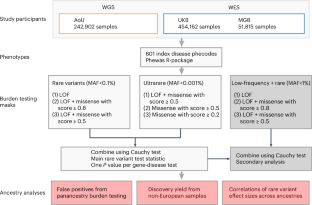
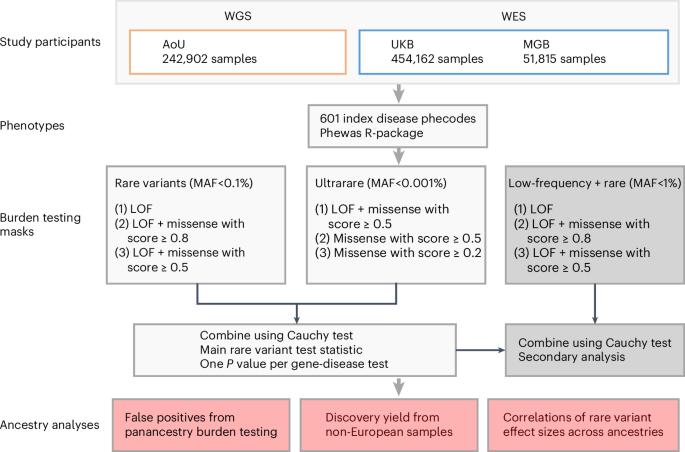
Large-scale sequencing has enabled unparalleled opportunities to investigate the role of rare coding variation in human phenotypic variability. Here, we present a pan-ancestry analysis of sequencing data from three large biobanks, including the All of Us research program. Using mixed-effects models, we performed gene-based rare variant testing for 601 diseases across 748,879 individuals, including 155,236 with ancestry dissimilar to European. We identified 363 significant associations, which highlighted core genes for the human disease phenome and identified potential novel associations, including UBR3 for cardiometabolic disease and YLPM1 for psychiatric disease. Pan-ancestry burden testing represented an inclusive and useful approach for discovery in diverse datasets, although we also highlight the importance of ancestry-specific sensitivity analyses in this setting. Finally, we found that effect sizes for rare protein-disrupting variants were concordant between samples similar to European ancestry and other genetic ancestries (βDeming = 0.7–1.0). Our results have implications for multi-ancestry and cross-biobank approaches in sequencing association studies for human disease. Gene-based rare variant analyses for 601 diseases across 748,879 individuals from three biobanks identify 363 significant associations and highlight important considerations for multi-ancestry and cross-biobank sequencing studies.
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
