{"title":"Predicting CO2 equilibrium solubility in various amine-CO2 systems using an artificial neural network model","authors":"Apri Wahyudi , Uthaiporn Suriyapraphadilok","doi":"10.1016/j.egyai.2024.100426","DOIUrl":null,"url":null,"abstract":"<div><p>Three proposed reaction mechanisms can occur in an amine-CO<sub>2</sub> system: either zwitterionic or termolecular mechanisms for primary/secondary amines and base-catalyzed hydration for tertiary amines. The intricacy of this system hinders the construction of a general model for all types of amines. This study attempts to build an artificial neural network model that predicts the equilibrium solubility of any nonblended aqueous amine-CO<sub>2</sub> system under given operating conditions, regardless of the reaction mechanism. This is a novel approach that has not yet been reported. The amines were characterized using molecular descriptors derived from COSMO theory through density functional theory calculations to incorporate molecular structures as model features. Our model achieved performance metrics (R<sup>2</sup>) of 0.9645 and 0.9481 for the training and validation sets, respectively. For unfamiliar amines that were absent in both the training and validation sets, our model achieved an R<sup>2</sup> of 0.8601. Model benchmarking was performed using a previously established thermodynamic model. Interpretations of the model are also provided based on the chosen features. This study also offers exploratory insight into how the molecular structure and operating conditions affect the CO<sub>2</sub> equilibrium solubility in amines. The model developed in this study has the potential to reduce the solvent screening time in determining appropriate amines for larger-scale applications.</p></div>","PeriodicalId":34138,"journal":{"name":"Energy and AI","volume":"18 ","pages":"Article 100426"},"PeriodicalIF":9.6000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2666546824000922/pdfft?md5=647dc9192891d787388e9c313289c368&pid=1-s2.0-S2666546824000922-main.pdf","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Energy and AI","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2666546824000922","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/16 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"COMPUTER SCIENCE, ARTIFICIAL INTELLIGENCE","Score":null,"Total":0}
引用次数: 0
Abstract
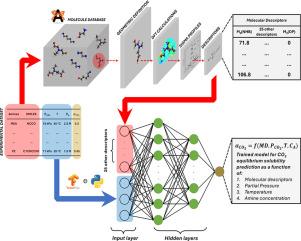
Three proposed reaction mechanisms can occur in an amine-CO2 system: either zwitterionic or termolecular mechanisms for primary/secondary amines and base-catalyzed hydration for tertiary amines. The intricacy of this system hinders the construction of a general model for all types of amines. This study attempts to build an artificial neural network model that predicts the equilibrium solubility of any nonblended aqueous amine-CO2 system under given operating conditions, regardless of the reaction mechanism. This is a novel approach that has not yet been reported. The amines were characterized using molecular descriptors derived from COSMO theory through density functional theory calculations to incorporate molecular structures as model features. Our model achieved performance metrics (R2) of 0.9645 and 0.9481 for the training and validation sets, respectively. For unfamiliar amines that were absent in both the training and validation sets, our model achieved an R2 of 0.8601. Model benchmarking was performed using a previously established thermodynamic model. Interpretations of the model are also provided based on the chosen features. This study also offers exploratory insight into how the molecular structure and operating conditions affect the CO2 equilibrium solubility in amines. The model developed in this study has the potential to reduce the solvent screening time in determining appropriate amines for larger-scale applications.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
