{"title":"Quantum Chemical Molecular Dynamics Simulations for Methane-Water Cages","authors":"Giuseppe Lanza","doi":"10.1002/qua.27487","DOIUrl":null,"url":null,"abstract":"<p>The dynamic stability of various methane-water clathrate-like cages (CH<sub>4</sub>@(H<sub>2</sub>O)<sub><i>n</i></sub>, <i>n</i> = 16, 18, 20, 22), has been analyzed explicitly considering thermal effects by means of ab initio M06-2X/6–31+G*/PCM calculations, which make use of Gaussian basis functions. Starting from the equilibrium filled cage structures, <i>classical</i>, dynamic reaction coordinate (DRC) on the Born–Oppenheimer surface, and <i>semiclassical</i>, Born–Oppenheimer plus harmonic zero-point energy surface (BOMD), molecular dynamics have been carried out. Water molecules have a high tendency to orient covalent O–H bonds tangentially to the hydrophobic surface, thus clathrate-like arrangements are an acceptable model to fully hydrate methane. If the cage size is such as to minimize core repulsion, due to electron cloud overlap, and to maximize host–guest van der Waals attractions, the clathrate-like structures have a life-time of two picoseconds in <i>classical</i> DRC simulations. The inclusion of quantum kinetic energy in BOMD simulations results in less structured cages with a reduced amount of hydrogen bond network. The preferential tangential orientation of the O-H bonds is largely maintained, although few of them point toward the methane for a very short time in BOMD simulations. The reduced configurational space of water molecules hydrating hydrophobic moiety is highlighted, thus any satisfactory molecular modeling has to account for it.</p>","PeriodicalId":182,"journal":{"name":"International Journal of Quantum Chemistry","volume":"124 19","pages":""},"PeriodicalIF":2.0000,"publicationDate":"2024-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/qua.27487","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Quantum Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/qua.27487","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
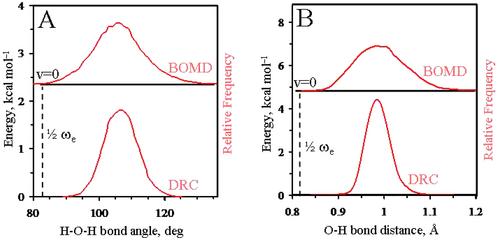
The dynamic stability of various methane-water clathrate-like cages (CH4@(H2O)n, n = 16, 18, 20, 22), has been analyzed explicitly considering thermal effects by means of ab initio M06-2X/6–31+G*/PCM calculations, which make use of Gaussian basis functions. Starting from the equilibrium filled cage structures, classical, dynamic reaction coordinate (DRC) on the Born–Oppenheimer surface, and semiclassical, Born–Oppenheimer plus harmonic zero-point energy surface (BOMD), molecular dynamics have been carried out. Water molecules have a high tendency to orient covalent O–H bonds tangentially to the hydrophobic surface, thus clathrate-like arrangements are an acceptable model to fully hydrate methane. If the cage size is such as to minimize core repulsion, due to electron cloud overlap, and to maximize host–guest van der Waals attractions, the clathrate-like structures have a life-time of two picoseconds in classical DRC simulations. The inclusion of quantum kinetic energy in BOMD simulations results in less structured cages with a reduced amount of hydrogen bond network. The preferential tangential orientation of the O-H bonds is largely maintained, although few of them point toward the methane for a very short time in BOMD simulations. The reduced configurational space of water molecules hydrating hydrophobic moiety is highlighted, thus any satisfactory molecular modeling has to account for it.
期刊介绍:
Since its first formulation quantum chemistry has provided the conceptual and terminological framework necessary to understand atoms, molecules and the condensed matter. Over the past decades synergistic advances in the methodological developments, software and hardware have transformed quantum chemistry in a truly interdisciplinary science that has expanded beyond its traditional core of molecular sciences to fields as diverse as chemistry and catalysis, biophysics, nanotechnology and material science.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
