Inflammation-induced fever depends on prostaglandin E2 production by brain endothelial cells and EP3 receptors in the median preoptic nucleus of the hypothalamus
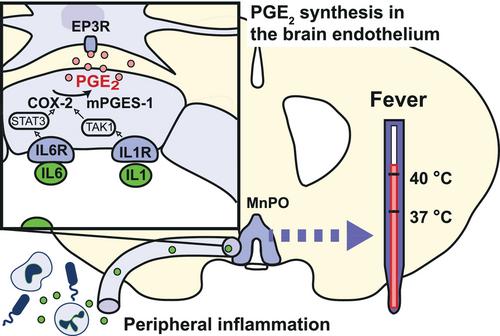
{"title":"Inflammation-induced fever depends on prostaglandin E2 production by brain endothelial cells and EP3 receptors in the median preoptic nucleus of the hypothalamus","authors":"Anders Blomqvist","doi":"10.1111/apha.14238","DOIUrl":null,"url":null,"abstract":"<p>I read with interest the editorial by Bai<span><sup>1</sup></span> on the paper by Yu et al.<span><sup>2</sup></span> on the role of caspase 11 in fever. However, I feel that the author ignores the absolutely critical role that prostaglandin (PG) E<sub>2</sub> production in brain endothelial cells has in generating fever, but rather seems to regard it as an auxiliary mechanism. Although both peripheral and central cytokine production may contribute to fever, as suggested by the study by Yu et al.,<span><sup>2</sup></span> the critical mechanism is PGE<sub>2</sub> synthesis and its binding to EP<sub>3</sub> receptor expressing neurons in the median preoptic nucleus (MnPO) of the hypothalamus.<span><sup>3, 4</sup></span> If PGE<sub>2</sub> synthesis is blocked or EP<sub>3</sub> receptors are deleted in the MnPO, no fever occurs,<span><sup>5, 6</sup></span> even though there still is increased cytokine production in the periphery and in the brain.<span><sup>7</sup></span> The critical PGE<sub>2</sub> synthesis occurs in brain endothelial cells as shown by the absence of fever when the PGE<sub>2</sub> synthesizing enzymes cyclooxygenase-2 (Cox-2) and microsomal prostaglandin E synthase-1 (mPGES-1) are deleted from these cells.<span><sup>8</sup></span> Cox-2 and mPGES-1 are in turn induced by cytokine binding to receptors on the endothelial cells<span><sup>9-11</sup></span> (Figure 1). If these receptors, such as those for IL-1 and IL-6, or their downstream signaling molecules are selectively deleted from brain endothelial cells, the fever is suppressed.<span><sup>13-16</sup></span></p><p>It should also be pointed out that the evidence for the involvement of microglial cells in inflammation-induced sickness responses, and in particular in fever, is far from clear. Although it is well recognized that peripheral inflammation activates microglial cells,<span><sup>17</sup></span> the mechanism behind this activation is not fully understood. It is unlikely due to direct action of cytokines on the microglial cells, particularly when it comes to interleukin-1, which is a major pyrogen,<span><sup>18</sup></span> because if transport across the blood–brain barrier at all occurs in any significant amount, microglial cells express negligible levels of IL-1 receptors.<span><sup>19</sup></span> The critical IL-1 receptor-expressing cells for IL-1 activation of microglial cells are the endothelial cells, which via an as-yet-unidentified messenger molecule by a paracrine mechanism activate the microglial cells.<span><sup>20</sup></span></p><p>While it is generally assumed that various sickness symptoms and neuropsychiatric disorders are associated with activated microglia,<span><sup>21</sup></span> apart from a study demonstrating a role of striatal microglial cells in negative affect elicited by peripheral inflammation,<span><sup>22</sup></span> there is very little evidence for a causal relationship between these phenomena. It is not even clear which brain cells are responsible for the increased levels of cytokines seen after peripheral inflammation. When microglial cells were depleted, LPS-induced cytokine expression in the brain was unaffected, as were disease symptoms such as body weight loss and suppressed motor activity.<span><sup>23</sup></span> The role of activated microglia in fever has not, to my knowledge, been investigated before.</p><p>Although the study by Yu et al.<span><sup>2</sup></span> suggests that microglial cells contribute to the fever response, there are several caveats that need to be considered. In one set of experiments, the authors used intracerebral injection of clodronate to delete microglial cells. However, clodronate also appears to target perivascular macrophages, as was shown by Schiltz and Sawchenko,<span><sup>24</sup></span> and while the clodronate injection reduced the response to peripheral injection of IL-1, as shown in a subsequent study from the Sawchenko laboratory,<span><sup>25</sup></span> it enhanced the response, including fever, to peripheral injection of LPS, that is, a finding opposite to that reported by Yu et al.<span><sup>2</sup></span> In another set of experiments, Yu et al. injected into the preoptic region an adenovirus expressing shRNA to silence the caspase 11 expression through RNA interference. Although they report reduced caspase 11 expression in microglia but not in neurons (and associated attenuated fever), they do not seem to have examined the extent to which caspase 11 expression was attenuated in other cell types such as perivascular macrophages and endothelial cells.</p><p>In conclusion, I feel that the findings by Yu et al.<span><sup>2</sup></span> and in particular, the idea the febrile response is enhanced by preoptic microglia, although interesting, should be interpreted with caution. Importantly, even if there were indeed such an enhancement of the fever signal, according to the evidence available today, such an enhanced signal would still need to be converted into PGE<sub>2</sub> synthesis by brain endothelial cells in order to augment the fever response. It is in this context of interest to note in the study by Yu et al.<span><sup>2</sup></span> that both the intracerebral chlodronate injection to deplete microglia and the RNA interference to silence caspase 11 were reported to result in decreased concentrations of PGE<sub>2</sub> in the brain, implying that the mechanism described indeed would be upstream of PGE<sub>2</sub> synthesis.</p><p>Anders Blomqvist: Writing – original draft; conceptualization.</p>","PeriodicalId":107,"journal":{"name":"Acta Physiologica","volume":"240 12","pages":""},"PeriodicalIF":5.6000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/apha.14238","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Physiologica","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/apha.14238","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PHYSIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
I read with interest the editorial by Bai1 on the paper by Yu et al.2 on the role of caspase 11 in fever. However, I feel that the author ignores the absolutely critical role that prostaglandin (PG) E2 production in brain endothelial cells has in generating fever, but rather seems to regard it as an auxiliary mechanism. Although both peripheral and central cytokine production may contribute to fever, as suggested by the study by Yu et al.,2 the critical mechanism is PGE2 synthesis and its binding to EP3 receptor expressing neurons in the median preoptic nucleus (MnPO) of the hypothalamus.3, 4 If PGE2 synthesis is blocked or EP3 receptors are deleted in the MnPO, no fever occurs,5, 6 even though there still is increased cytokine production in the periphery and in the brain.7 The critical PGE2 synthesis occurs in brain endothelial cells as shown by the absence of fever when the PGE2 synthesizing enzymes cyclooxygenase-2 (Cox-2) and microsomal prostaglandin E synthase-1 (mPGES-1) are deleted from these cells.8 Cox-2 and mPGES-1 are in turn induced by cytokine binding to receptors on the endothelial cells9-11 (Figure 1). If these receptors, such as those for IL-1 and IL-6, or their downstream signaling molecules are selectively deleted from brain endothelial cells, the fever is suppressed.13-16
It should also be pointed out that the evidence for the involvement of microglial cells in inflammation-induced sickness responses, and in particular in fever, is far from clear. Although it is well recognized that peripheral inflammation activates microglial cells,17 the mechanism behind this activation is not fully understood. It is unlikely due to direct action of cytokines on the microglial cells, particularly when it comes to interleukin-1, which is a major pyrogen,18 because if transport across the blood–brain barrier at all occurs in any significant amount, microglial cells express negligible levels of IL-1 receptors.19 The critical IL-1 receptor-expressing cells for IL-1 activation of microglial cells are the endothelial cells, which via an as-yet-unidentified messenger molecule by a paracrine mechanism activate the microglial cells.20
While it is generally assumed that various sickness symptoms and neuropsychiatric disorders are associated with activated microglia,21 apart from a study demonstrating a role of striatal microglial cells in negative affect elicited by peripheral inflammation,22 there is very little evidence for a causal relationship between these phenomena. It is not even clear which brain cells are responsible for the increased levels of cytokines seen after peripheral inflammation. When microglial cells were depleted, LPS-induced cytokine expression in the brain was unaffected, as were disease symptoms such as body weight loss and suppressed motor activity.23 The role of activated microglia in fever has not, to my knowledge, been investigated before.
Although the study by Yu et al.2 suggests that microglial cells contribute to the fever response, there are several caveats that need to be considered. In one set of experiments, the authors used intracerebral injection of clodronate to delete microglial cells. However, clodronate also appears to target perivascular macrophages, as was shown by Schiltz and Sawchenko,24 and while the clodronate injection reduced the response to peripheral injection of IL-1, as shown in a subsequent study from the Sawchenko laboratory,25 it enhanced the response, including fever, to peripheral injection of LPS, that is, a finding opposite to that reported by Yu et al.2 In another set of experiments, Yu et al. injected into the preoptic region an adenovirus expressing shRNA to silence the caspase 11 expression through RNA interference. Although they report reduced caspase 11 expression in microglia but not in neurons (and associated attenuated fever), they do not seem to have examined the extent to which caspase 11 expression was attenuated in other cell types such as perivascular macrophages and endothelial cells.
In conclusion, I feel that the findings by Yu et al.2 and in particular, the idea the febrile response is enhanced by preoptic microglia, although interesting, should be interpreted with caution. Importantly, even if there were indeed such an enhancement of the fever signal, according to the evidence available today, such an enhanced signal would still need to be converted into PGE2 synthesis by brain endothelial cells in order to augment the fever response. It is in this context of interest to note in the study by Yu et al.2 that both the intracerebral chlodronate injection to deplete microglia and the RNA interference to silence caspase 11 were reported to result in decreased concentrations of PGE2 in the brain, implying that the mechanism described indeed would be upstream of PGE2 synthesis.
Anders Blomqvist: Writing – original draft; conceptualization.
期刊介绍:
Acta Physiologica is an important forum for the publication of high quality original research in physiology and related areas by authors from all over the world. Acta Physiologica is a leading journal in human/translational physiology while promoting all aspects of the science of physiology. The journal publishes full length original articles on important new observations as well as reviews and commentaries.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
