{"title":"Case Study: Analyzing CFTR Mutations and SNPs in Pulmonary Fibrosis Patients with Unclear Symptoms.","authors":"Sahar Yousaf, Sumaira, Iqbal Bano, Atia Rehman, Samra Kousar, Muhammad Usman Ghani, Mariam Shahid","doi":"10.1155/2024/8836342","DOIUrl":null,"url":null,"abstract":"<p><p>Cystic fibrosis (CF) is a genetic monogenic disorder inherited in an autosomal recessive manner, marked by persistent airway infections in the endobronchial region. This condition leads to the gradual development of bronchiectasis and, ultimately, respiratory failure, emerging as the primary cause of mortality in individuals diagnosed with CF. Diagnosis is done depending on the patient's symptoms and lung radiological findings like chest X-rays and CTs. In younger patients and children, diagnosis becomes difficult due to overlapping symptoms with other diseases such as CF which is a rare genetic disease in our population. Diagnosis of CF usually relies on characteristic symptoms, a family history of CF, and an abnormal sweat chloride test, but in children, low sweat production during testing leads to false negative results. In this case report, a suspected patient with ambiguous respiratory symptoms underwent a comprehensive investigation revealing elevated CRP levels, TLC, and characteristic pulmonary manifestations on chest X-ray, suggesting cystic fibrosis. Despite negative sweat chloride tests, the patient was analysed for potential candidate SNPs and was also tested for potential CFTR mutations to rule out CF, genetic analysis confirmed the diagnosis. Genetic testing plays a crucial role in diagnosing cystic fibrosis, even when traditional tests are inconclusive. Specific mutations like Δ508 deletion and rs213950 guide personalized treatment. Consanguinity and family history highlight genetic predisposition, while environmental factors may influence symptom onset. Further research is needed to understand these complexities and improve diagnostic and treatment approaches.</p>","PeriodicalId":9627,"journal":{"name":"Case Reports in Medicine","volume":"2024 ","pages":"8836342"},"PeriodicalIF":0.7000,"publicationDate":"2024-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11442034/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2024/8836342","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
引用次数: 0
Abstract
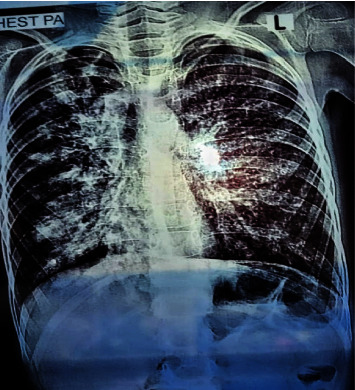
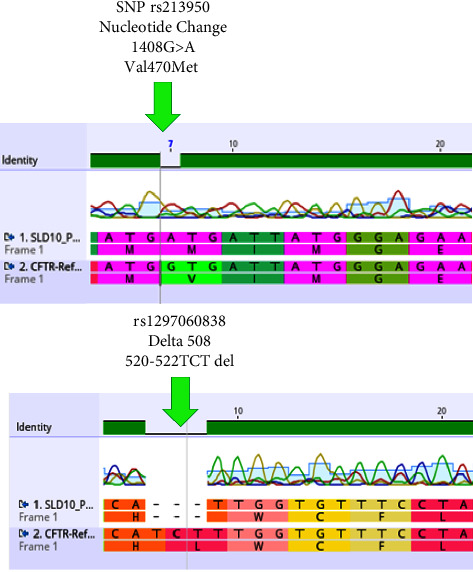
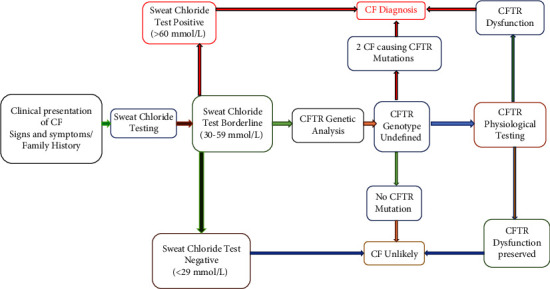
Cystic fibrosis (CF) is a genetic monogenic disorder inherited in an autosomal recessive manner, marked by persistent airway infections in the endobronchial region. This condition leads to the gradual development of bronchiectasis and, ultimately, respiratory failure, emerging as the primary cause of mortality in individuals diagnosed with CF. Diagnosis is done depending on the patient's symptoms and lung radiological findings like chest X-rays and CTs. In younger patients and children, diagnosis becomes difficult due to overlapping symptoms with other diseases such as CF which is a rare genetic disease in our population. Diagnosis of CF usually relies on characteristic symptoms, a family history of CF, and an abnormal sweat chloride test, but in children, low sweat production during testing leads to false negative results. In this case report, a suspected patient with ambiguous respiratory symptoms underwent a comprehensive investigation revealing elevated CRP levels, TLC, and characteristic pulmonary manifestations on chest X-ray, suggesting cystic fibrosis. Despite negative sweat chloride tests, the patient was analysed for potential candidate SNPs and was also tested for potential CFTR mutations to rule out CF, genetic analysis confirmed the diagnosis. Genetic testing plays a crucial role in diagnosing cystic fibrosis, even when traditional tests are inconclusive. Specific mutations like Δ508 deletion and rs213950 guide personalized treatment. Consanguinity and family history highlight genetic predisposition, while environmental factors may influence symptom onset. Further research is needed to understand these complexities and improve diagnostic and treatment approaches.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
