Jonathan S Van Buskirk, Gordon G C Peterson, Daniel C Fredrickson
{"title":"Machine Learning-Based Investigation of Atomic Packing Effects: Chemical Pressures at the Extremes of Intermetallic Complexity.","authors":"Jonathan S Van Buskirk, Gordon G C Peterson, Daniel C Fredrickson","doi":"10.1021/jacs.4c10479","DOIUrl":null,"url":null,"abstract":"<p><p>Intermetallic phases represent a domain of emergent behavior, in which atoms with packing and electronic preferences can combine into complex geometrical arrangements whose long-range order involves repeat patterns containing thousands of atoms or is incompatible with a 3D unit cell. The formation of such arrangements points to unexplained driving forces within these systems that, if understood, could be harnessed in the design of new metallic materials. DFT-chemical pressure (CP) analysis has emerged as an approach to visualize how atomic packing tensions within simpler crystal structures can drive this complexity and create potential functionality. However, the applications of this method have hitherto been limited in scope by its dependence on resource-intensive electronic structure calculations. In this Article, we develop machine learning (ML)-based implementation of the CP approach, drawing on the collection of DFT-CP schemes in the Intermetallic Reactivity Database. We illustrate the method with comparisons of ML-CP and DFT-CP schemes for a series of examples, before demonstrating its application with an exploration of one of the quintessential instances of complexity in intermetallic chemistry, Mg<sub>2</sub>Al<sub>3</sub>, whose high-temperature unit cell is a 2.8 nm cube containing 1227 atoms. An analysis of its ML-CP-derived interatomic pressures traces the origins of the structure to simple matching rules for the assembly of Frank-Kasper polyhedra. The ML-CP model can be immediately employed on other intermetallic systems, through either its web interface or a command-line version, with just a crystallographic information file.</p>","PeriodicalId":49,"journal":{"name":"Journal of the American Chemical Society","volume":" ","pages":""},"PeriodicalIF":15.6000,"publicationDate":"2024-10-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the American Chemical Society","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/jacs.4c10479","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
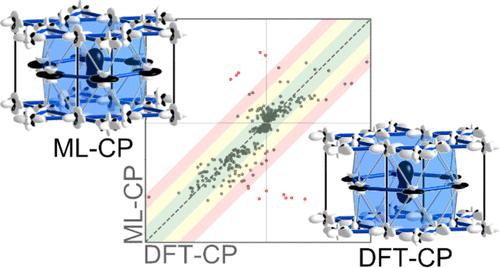
Intermetallic phases represent a domain of emergent behavior, in which atoms with packing and electronic preferences can combine into complex geometrical arrangements whose long-range order involves repeat patterns containing thousands of atoms or is incompatible with a 3D unit cell. The formation of such arrangements points to unexplained driving forces within these systems that, if understood, could be harnessed in the design of new metallic materials. DFT-chemical pressure (CP) analysis has emerged as an approach to visualize how atomic packing tensions within simpler crystal structures can drive this complexity and create potential functionality. However, the applications of this method have hitherto been limited in scope by its dependence on resource-intensive electronic structure calculations. In this Article, we develop machine learning (ML)-based implementation of the CP approach, drawing on the collection of DFT-CP schemes in the Intermetallic Reactivity Database. We illustrate the method with comparisons of ML-CP and DFT-CP schemes for a series of examples, before demonstrating its application with an exploration of one of the quintessential instances of complexity in intermetallic chemistry, Mg2Al3, whose high-temperature unit cell is a 2.8 nm cube containing 1227 atoms. An analysis of its ML-CP-derived interatomic pressures traces the origins of the structure to simple matching rules for the assembly of Frank-Kasper polyhedra. The ML-CP model can be immediately employed on other intermetallic systems, through either its web interface or a command-line version, with just a crystallographic information file.
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
