{"title":"A Novel Ensemble Approach with Deep Transfer Learning for Accurate Identification of Foodborne Bacteria from Hyperspectral Microscopy","authors":"Qurrat ul Ain , Sohaib Asif","doi":"10.1016/j.compbiolchem.2024.108238","DOIUrl":null,"url":null,"abstract":"<div><div>The detection of foodborne bacteria is critical in ensuring both consumer safety and food safety. If these pathogens are not properly identified, it can lead to dangerous cross-contamination. One of the most common methods for classifying bacteria is through the examination of Hyperspectral microscope imaging (HMI). A widely used technique for measuring microbial growth is microscopic cell counting. HMI is a laborious and expensive process, producing voluminous data and needing specialized equipment, which might not be widely available. Machine learning (ML) methods are now frequently utilized to automatically interpret data from hyperspectral microscopy. The objective of our study is to devise a technique that employs deep transfer learning to address the challenge of limited data and utilizes four base classifiers - InceptionResNetV2, MobileNet, ResNet101V2, and Xception - to create an ensemble-based classification model for distinguishing live and dead bacterial cells of six pathogenic strains. In order to determine the optimal weights for the base classifiers, a Powell's optimization method was utilized in conjunction with a weighted average ensemble (WAVE) technique. We carried out an extensive experimental study to verify the efficiency of our proposed ensemble model on live and dead cell images of six different foodborne bacteria. In order to gain a better understanding of the regions, we performed a Grad-CAM analysis to explain the predictions made by our model. Through a series of experiments, our proposed framework has proven its capacity to effectively and precisely detect numerous bacterial pathogens. Specifically, it achieved a perfect identification rate of 100% for <em>Escherichia coli (EC), Listeria innocua (LI), and Salmonella Enteritidis (SE)</em>, while achieving rates of 96.30% for <em>Salmonella Typhimurium (ST),</em> 87.13% for <em>Staphylococcus aureus (SA</em>), and 94.12% for Salmonella Heidelberg (SH). As a result, it can be considered as an effective tool for the identification of foodborne pathogens, due to its high level of efficiency.</div></div>","PeriodicalId":10616,"journal":{"name":"Computational Biology and Chemistry","volume":"113 ","pages":"Article 108238"},"PeriodicalIF":3.1000,"publicationDate":"2024-10-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Biology and Chemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1476927124002263","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
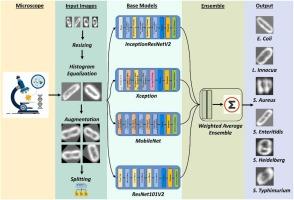
The detection of foodborne bacteria is critical in ensuring both consumer safety and food safety. If these pathogens are not properly identified, it can lead to dangerous cross-contamination. One of the most common methods for classifying bacteria is through the examination of Hyperspectral microscope imaging (HMI). A widely used technique for measuring microbial growth is microscopic cell counting. HMI is a laborious and expensive process, producing voluminous data and needing specialized equipment, which might not be widely available. Machine learning (ML) methods are now frequently utilized to automatically interpret data from hyperspectral microscopy. The objective of our study is to devise a technique that employs deep transfer learning to address the challenge of limited data and utilizes four base classifiers - InceptionResNetV2, MobileNet, ResNet101V2, and Xception - to create an ensemble-based classification model for distinguishing live and dead bacterial cells of six pathogenic strains. In order to determine the optimal weights for the base classifiers, a Powell's optimization method was utilized in conjunction with a weighted average ensemble (WAVE) technique. We carried out an extensive experimental study to verify the efficiency of our proposed ensemble model on live and dead cell images of six different foodborne bacteria. In order to gain a better understanding of the regions, we performed a Grad-CAM analysis to explain the predictions made by our model. Through a series of experiments, our proposed framework has proven its capacity to effectively and precisely detect numerous bacterial pathogens. Specifically, it achieved a perfect identification rate of 100% for Escherichia coli (EC), Listeria innocua (LI), and Salmonella Enteritidis (SE), while achieving rates of 96.30% for Salmonella Typhimurium (ST), 87.13% for Staphylococcus aureus (SA), and 94.12% for Salmonella Heidelberg (SH). As a result, it can be considered as an effective tool for the identification of foodborne pathogens, due to its high level of efficiency.
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
