{"title":"Autoencoder-based drug synergy framework for malignant diseases","authors":"Pooja Rani , Kamlesh Dutta , Vijay Kumar","doi":"10.1016/j.compbiolchem.2024.108273","DOIUrl":null,"url":null,"abstract":"<div><div>Drug combination emerges as a viable option for the treatment of malignant diseases. Drug combination outperforms monotherapy by improving therapeutic efficacy, reducing toxicity, and overcoming drug resistance. To find viable drug combinations it is difficult to traverse empirically because of enormous combinational space. Machine learning and deep learning approaches are used to uncover novel synergistic drug combinations in enormous combinational space. Here, AESyn, a novel autoencoder-based drug synergy framework for malignant diseases using a bag of words encoding is proposed. The bag of word encoding technique is used to extract drug-targeted genes. The framework utilized screening data from NCI-ALMANAC, and O’Neil datasets. Autoencoders take drug embeddings with drug-targeted genes as input for processing. The autoencoder in the proposed framework is used to extract drug features. The proposed framework is evaluated on classification and regression metrics. The performance of the proposed framework is compared with existing methods of drug synergy. According to the findings, the proposed framework achieved high performance with an accuracy of 95%, AUROC of 94.2%, and MAPE of 7.2. The autoencoder-based framework for malignant diseases using an encoding technique provides a stable, order-independent drug synergy prediction.</div></div>","PeriodicalId":10616,"journal":{"name":"Computational Biology and Chemistry","volume":"113 ","pages":"Article 108273"},"PeriodicalIF":3.1000,"publicationDate":"2024-11-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational Biology and Chemistry","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1476927124002615","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
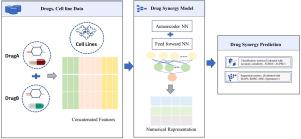
Drug combination emerges as a viable option for the treatment of malignant diseases. Drug combination outperforms monotherapy by improving therapeutic efficacy, reducing toxicity, and overcoming drug resistance. To find viable drug combinations it is difficult to traverse empirically because of enormous combinational space. Machine learning and deep learning approaches are used to uncover novel synergistic drug combinations in enormous combinational space. Here, AESyn, a novel autoencoder-based drug synergy framework for malignant diseases using a bag of words encoding is proposed. The bag of word encoding technique is used to extract drug-targeted genes. The framework utilized screening data from NCI-ALMANAC, and O’Neil datasets. Autoencoders take drug embeddings with drug-targeted genes as input for processing. The autoencoder in the proposed framework is used to extract drug features. The proposed framework is evaluated on classification and regression metrics. The performance of the proposed framework is compared with existing methods of drug synergy. According to the findings, the proposed framework achieved high performance with an accuracy of 95%, AUROC of 94.2%, and MAPE of 7.2. The autoencoder-based framework for malignant diseases using an encoding technique provides a stable, order-independent drug synergy prediction.
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
