Changes in Plasma Clearance of CYP450 Probe Drugs May Not be Specific for Altered In Vivo Enzyme Activity Under (Patho)Physiological Conditions: How to Interpret Findings of Probe Cocktail Studies.
Laura M de Jong, Marinda van de Kreeke, Mariam Ahmadi, Jesse J Swen, Catherijne A J Knibbe, J G Coen van Hasselt, Martijn L Manson, Elke H J Krekels
{"title":"Changes in Plasma Clearance of CYP450 Probe Drugs May Not be Specific for Altered In Vivo Enzyme Activity Under (Patho)Physiological Conditions: How to Interpret Findings of Probe Cocktail Studies.","authors":"Laura M de Jong, Marinda van de Kreeke, Mariam Ahmadi, Jesse J Swen, Catherijne A J Knibbe, J G Coen van Hasselt, Martijn L Manson, Elke H J Krekels","doi":"10.1007/s40262-024-01426-8","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objective: </strong>CYP450 (CYP) phenotyping involves quantifying an individual's plasma clearance of CYP-specific probe drugs, as a proxy for in vivo CYP enzyme activity. It is increasingly applied to study alterations in CYP enzyme activity under various (patho)physiological conditions, such as inflammation, obesity, or pregnancy. The phenotyping approach assumes that changes in plasma clearance of probe drugs are driven by changes in CYP enzyme activity. However, plasma clearance is also influenced by protein binding, blood-to-plasma ratio, and hepatic blood flow, all of which may change under (patho)physiological conditions.</p><p><strong>Methods: </strong>Using a physiologically based pharmacokinetic (PBPK) workflow, we aimed to evaluate whether the plasma clearance of commonly used CYP probe drugs is indeed directly proportional to alterations in CYP enzyme activity (sensitivity), and to what extent alterations in protein binding, blood-to-plasma ratio, and hepatic blood flow observed under (patho)physiological conditions impact plasma clearance (specificity).</p><p><strong>Results: </strong>Plasma clearance of CYP probe drugs is sensitive to alterations in CYP enzyme activity, since alterations in intrinsic clearance between - 90% and + 150% resulted in near-proportional changes in plasma clearance, except for midazolam in the case of > 50% CYP3A4 induction. However, plasma clearance also changed near-proportionally with alterations in the unbound drug fraction, diminishing probe specificity. This was particularly relevant for high protein-bound probe drugs, as alterations in plasma protein binding resulted in larger relative changes in the unbound drug fraction. Alterations in the blood-to-plasma ratio and hepatic blood flow of ± 50% resulted in plasma clearance changes of less than ± 16%, meaning they limitedly impacted plasma clearance of CYP probe drugs, except for midazolam. In order to correct for the impact of non-metabolic determinants on probe drug plasma clearance, an R script was developed to calculate how much the CYP enzyme activity is actually altered under (patho)physiological conditions, when alterations in the unbound drug fraction, blood-to-plasma ratio, and/or hepatic blood flow also impact probe drug plasma clearance.</p><p><strong>Conclusions: </strong>As plasma protein binding can change under (patho)physiological conditions, alterations in unbound drug fraction should be accounted for when using CYP probe drug plasma clearance as a proxy for CYP enzyme activity in patient populations. The tool developed in this study can support researchers in determining alterations in CYP enzyme activity in patients with (patho)physiological conditions.</p>","PeriodicalId":10405,"journal":{"name":"Clinical Pharmacokinetics","volume":" ","pages":"1585-1595"},"PeriodicalIF":4.0000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11573838/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pharmacokinetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s40262-024-01426-8","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/27 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 0
Abstract
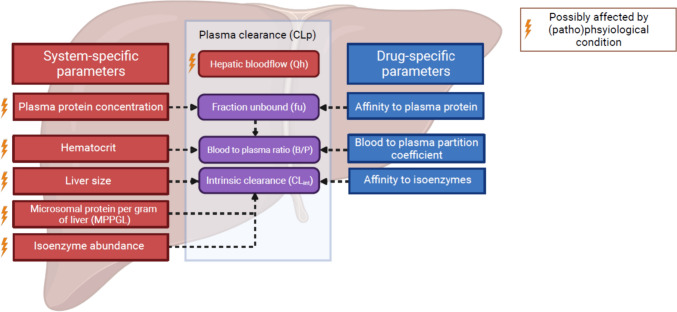
Background and objective: CYP450 (CYP) phenotyping involves quantifying an individual's plasma clearance of CYP-specific probe drugs, as a proxy for in vivo CYP enzyme activity. It is increasingly applied to study alterations in CYP enzyme activity under various (patho)physiological conditions, such as inflammation, obesity, or pregnancy. The phenotyping approach assumes that changes in plasma clearance of probe drugs are driven by changes in CYP enzyme activity. However, plasma clearance is also influenced by protein binding, blood-to-plasma ratio, and hepatic blood flow, all of which may change under (patho)physiological conditions.
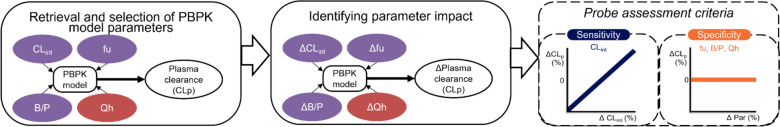
Methods: Using a physiologically based pharmacokinetic (PBPK) workflow, we aimed to evaluate whether the plasma clearance of commonly used CYP probe drugs is indeed directly proportional to alterations in CYP enzyme activity (sensitivity), and to what extent alterations in protein binding, blood-to-plasma ratio, and hepatic blood flow observed under (patho)physiological conditions impact plasma clearance (specificity).
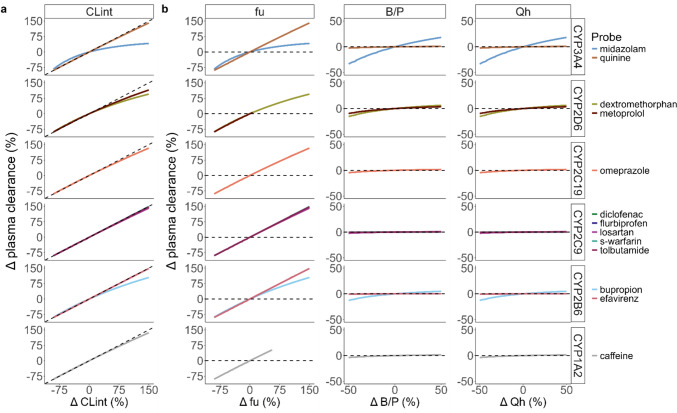
Results: Plasma clearance of CYP probe drugs is sensitive to alterations in CYP enzyme activity, since alterations in intrinsic clearance between - 90% and + 150% resulted in near-proportional changes in plasma clearance, except for midazolam in the case of > 50% CYP3A4 induction. However, plasma clearance also changed near-proportionally with alterations in the unbound drug fraction, diminishing probe specificity. This was particularly relevant for high protein-bound probe drugs, as alterations in plasma protein binding resulted in larger relative changes in the unbound drug fraction. Alterations in the blood-to-plasma ratio and hepatic blood flow of ± 50% resulted in plasma clearance changes of less than ± 16%, meaning they limitedly impacted plasma clearance of CYP probe drugs, except for midazolam. In order to correct for the impact of non-metabolic determinants on probe drug plasma clearance, an R script was developed to calculate how much the CYP enzyme activity is actually altered under (patho)physiological conditions, when alterations in the unbound drug fraction, blood-to-plasma ratio, and/or hepatic blood flow also impact probe drug plasma clearance.
Conclusions: As plasma protein binding can change under (patho)physiological conditions, alterations in unbound drug fraction should be accounted for when using CYP probe drug plasma clearance as a proxy for CYP enzyme activity in patient populations. The tool developed in this study can support researchers in determining alterations in CYP enzyme activity in patients with (patho)physiological conditions.
期刊介绍:
Clinical Pharmacokinetics promotes the continuing development of clinical pharmacokinetics and pharmacodynamics for the improvement of drug therapy, and for furthering postgraduate education in clinical pharmacology and therapeutics.
Pharmacokinetics, the study of drug disposition in the body, is an integral part of drug development and rational use. Knowledge and application of pharmacokinetic principles leads to accelerated drug development, cost effective drug use and a reduced frequency of adverse effects and drug interactions.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
