{"title":"Mechanistic studies on phosphine-catalyzed [4 + 3] annulation of β′-acetoxy allenoate with 1C,3N-dinucleophile","authors":"Kui Yuan, Hao-Ran Yang, Yang Wang","doi":"10.1016/j.comptc.2024.114924","DOIUrl":null,"url":null,"abstract":"<div><div>The mechanism and role of catalyst on the PPh<sub>3</sub>-catalyzed [4 + 3] annulation reaction have been systematically investigated using density functional theory (DFT) method. Based on the calculations, the possible mechanism contains six steps: nucleophilic addition of PPh<sub>3</sub> to allenoate to give Z-configured intermediate, cleavage of C<img>O bond for forming phosphonium diene, nucleophilic addition of phosphonium diene with anionic 1C,3N-dinucleophile, intramolecular <span><span>[1]</span></span>, <span><span>[5]</span></span>-proton shift, ring-closure, and dissociation of catalyst. Non-covalent interaction (NCI) analysis shows that the O<strong>⋯</strong>P interaction would be the key for leading to the Z-configured pathway more favorable and electron localization function (ELF) analysis indicates that the implication of PPh<sub>3</sub> can significantly lower the energy barrier involved in C<img>O cleavage process, which mainly because the addition of PPh<sub>3</sub> reduces the electron density of C<img>O bond and thus facilities the cleavage of C<img>O bond. This theoretical study would provide some clues for understanding the role of catalyst in a catalytic reaction.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1241 ","pages":"Article 114924"},"PeriodicalIF":3.0000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004638","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/19 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
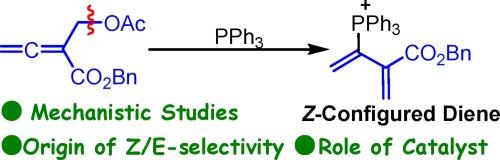
The mechanism and role of catalyst on the PPh3-catalyzed [4 + 3] annulation reaction have been systematically investigated using density functional theory (DFT) method. Based on the calculations, the possible mechanism contains six steps: nucleophilic addition of PPh3 to allenoate to give Z-configured intermediate, cleavage of CO bond for forming phosphonium diene, nucleophilic addition of phosphonium diene with anionic 1C,3N-dinucleophile, intramolecular [1], [5]-proton shift, ring-closure, and dissociation of catalyst. Non-covalent interaction (NCI) analysis shows that the O⋯P interaction would be the key for leading to the Z-configured pathway more favorable and electron localization function (ELF) analysis indicates that the implication of PPh3 can significantly lower the energy barrier involved in CO cleavage process, which mainly because the addition of PPh3 reduces the electron density of CO bond and thus facilities the cleavage of CO bond. This theoretical study would provide some clues for understanding the role of catalyst in a catalytic reaction.
利用密度泛函理论(DFT)方法系统地研究了 PPh3 催化[4 + 3]环化反应的机理和催化剂的作用。根据计算结果,可能的机理包括六个步骤:PPh3 与烯酸酯发生亲核加成反应,得到 Z 构型中间体;裂解 CO 键形成二烯膦;二烯膦与阴离子 1C,3N-亲核物发生亲核加成反应;分子内 [1]、[5]- 质子移动;闭环;催化剂解离。非共价相互作用(NCI)分析表明,O⋯P 相互作用是导致 Z 构型途径更有利的关键,而电子定位功能(ELF)分析表明,PPh3 的加入能显著降低 CO 裂解过程的能障,这主要是因为 PPh3 的加入降低了 CO 键的电子密度,从而促进了 CO 键的裂解。这项理论研究将为理解催化剂在催化反应中的作用提供一些线索。
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
