D.M. Tshwane , P.M. Maleka , R.S. Dima , L. Mogakane , T. Ngcobo , R.R. Maphanga
{"title":"Insights on formation of oxide layers, corrosion, and hydrogen embrittlement on the Ti2AlNb (1 1 0) surface: Density functional theory study","authors":"D.M. Tshwane , P.M. Maleka , R.S. Dima , L. Mogakane , T. Ngcobo , R.R. Maphanga","doi":"10.1016/j.comptc.2024.115002","DOIUrl":null,"url":null,"abstract":"<div><div>Ti<sub>2</sub>AlNb alloys offer good mechanical qualities and promise for use in various applications, such as aero-engines and other industries. However, corrosion and hydrogen embrittlement remain important concerns and limitations for their use. In this work, first-principle density functional theory is used to investigate the adsorption of hydrogen, fluorine and oxygen on the surface of Ti<sub>2</sub>AlNb (1<!--> <!-->1<!--> <!-->0). The effects of the considered adsorbates on the surface were compared by analysing the adsorption energy, charge density differences, density of states, and work function. The current findings revealed that the adsorption behaviour of all the adsorbates is exothermic and spontaneous due to the negative adsorption energy. More importantly, the effect of Van der Waals forces and dispersion correction was considered, it was found that for all adsorbates the dispersion correction approach exhibited the most stable adsorption energies (<span><math><mrow><msubsup><mi>E</mi><mrow><mi>ads</mi></mrow><mrow><mi>DFT</mi><mo>-</mo><mi>D</mi></mrow></msubsup></mrow></math></span>) than the standard density functional theory (<span><math><mrow><msubsup><mi>E</mi><mrow><mi>ads</mi></mrow><mrow><mi>DFT</mi></mrow></msubsup></mrow></math></span>). Thus, the standard DFT underestimates the adsorption energy. Furthermore, it was shown that the adsorption energy strength is dependent on the surface adsorption site, with the Ti-Nb and Al-Nb bridge sites being the most preferred sites for hydrogen, fluorine and oxygen adsorption. Subsequently, it was discovered that oxygen adsorption on the surface of Ti<sub>2</sub>AlNb (1<!--> <!-->1<!--> <!-->0) was more thermodynamically stable than hydrogen and fluorine. This suggests that the Ti<sub>2</sub>AlNb surface will likely suffer from oxidation rather than corrosion and hydrogen embrittlement. In addition, surface atoms showed electron-charge depletion, while adsorbates showed charge accumulation. The adsorption caused charge density redistribution and altered the surface work function.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1244 ","pages":"Article 115002"},"PeriodicalIF":2.9000,"publicationDate":"2025-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24005413","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/26 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
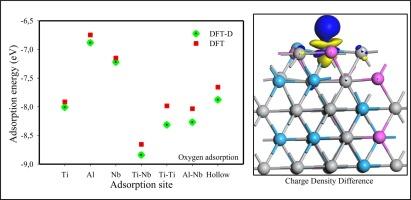
Ti2AlNb alloys offer good mechanical qualities and promise for use in various applications, such as aero-engines and other industries. However, corrosion and hydrogen embrittlement remain important concerns and limitations for their use. In this work, first-principle density functional theory is used to investigate the adsorption of hydrogen, fluorine and oxygen on the surface of Ti2AlNb (1 1 0). The effects of the considered adsorbates on the surface were compared by analysing the adsorption energy, charge density differences, density of states, and work function. The current findings revealed that the adsorption behaviour of all the adsorbates is exothermic and spontaneous due to the negative adsorption energy. More importantly, the effect of Van der Waals forces and dispersion correction was considered, it was found that for all adsorbates the dispersion correction approach exhibited the most stable adsorption energies () than the standard density functional theory (). Thus, the standard DFT underestimates the adsorption energy. Furthermore, it was shown that the adsorption energy strength is dependent on the surface adsorption site, with the Ti-Nb and Al-Nb bridge sites being the most preferred sites for hydrogen, fluorine and oxygen adsorption. Subsequently, it was discovered that oxygen adsorption on the surface of Ti2AlNb (1 1 0) was more thermodynamically stable than hydrogen and fluorine. This suggests that the Ti2AlNb surface will likely suffer from oxidation rather than corrosion and hydrogen embrittlement. In addition, surface atoms showed electron-charge depletion, while adsorbates showed charge accumulation. The adsorption caused charge density redistribution and altered the surface work function.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
