{"title":"Geometrical features, stability and hydrogen positions in (Al2Cu)n clusters","authors":"Xi Wang , Yule Yan , Qiman Liu","doi":"10.1016/j.comptc.2024.114999","DOIUrl":null,"url":null,"abstract":"<div><div>The phase equilibria of the Al-Cu alloys have been well-established, with the Al<sub>2</sub>Cu being a crucial component of the phase diagram. Constructing an atomic model with a 2:1 stoichiometric ratio of Al and Cu holds significance for further investigating the local structures of the alloy phases. Here, we employ the GA-DFT method to explore the structural potential energy surfaces of (Al<sub>2</sub>Cu)<sub>n</sub> clusters (n = 1–6). The results reveal that the (Al<sub>2</sub>Cu)<sub>n</sub> evolve from hollow cages to more densely packed configurations, with Al atoms relatively more concentrated and Cu atoms becoming more dispersed throughout the structures. The <em>E</em><sub>b</sub> and Δ<sub>2</sub><em>E</em> analyses show that the (Al<sub>2</sub>Cu)<sub>3</sub> has a higher stability than that of its neighbors, and the AIMD simulations demonstrate that it can maintain the structural integrity at 700 K. The molecular orbitals reveal that 21 valence electrons of the (Al<sub>2</sub>Cu)<sub>3</sub> fill superatomic orbits resulting in an electronic configuration of 1S<sup>2</sup>1P<sup>6</sup>1D<sup>10</sup>2S<sup>2</sup>1F<sup>1</sup>, which is also confirmed by the density of states. The good stability of the (Al<sub>2</sub>Cu)<sub>3</sub> allows the bonding of the H atom to it without causing significant deformation changes in the parent geometry, in which the H tends to preferentially locate at Al sites. The deformation of structures is particularly obvious when H is close to Cu atom.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1243 ","pages":"Article 114999"},"PeriodicalIF":3.0000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24005383","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/26 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
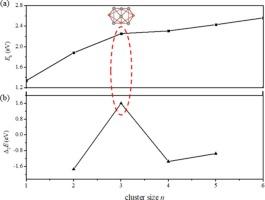
The phase equilibria of the Al-Cu alloys have been well-established, with the Al2Cu being a crucial component of the phase diagram. Constructing an atomic model with a 2:1 stoichiometric ratio of Al and Cu holds significance for further investigating the local structures of the alloy phases. Here, we employ the GA-DFT method to explore the structural potential energy surfaces of (Al2Cu)n clusters (n = 1–6). The results reveal that the (Al2Cu)n evolve from hollow cages to more densely packed configurations, with Al atoms relatively more concentrated and Cu atoms becoming more dispersed throughout the structures. The Eb and Δ2E analyses show that the (Al2Cu)3 has a higher stability than that of its neighbors, and the AIMD simulations demonstrate that it can maintain the structural integrity at 700 K. The molecular orbitals reveal that 21 valence electrons of the (Al2Cu)3 fill superatomic orbits resulting in an electronic configuration of 1S21P61D102S21F1, which is also confirmed by the density of states. The good stability of the (Al2Cu)3 allows the bonding of the H atom to it without causing significant deformation changes in the parent geometry, in which the H tends to preferentially locate at Al sites. The deformation of structures is particularly obvious when H is close to Cu atom.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
