Benjamin Ries, Richard J Gowers, Hannah M Baumann, David W H Swenson, Michael M Henry, James R B Eastwood, Irfan Alibay, David Mobley
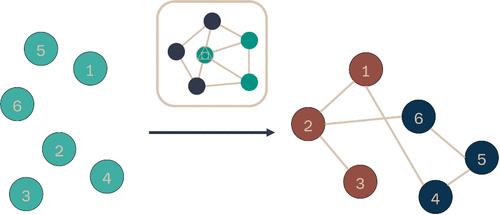
{"title":"Konnektor: A Framework for Using Graph Theory to Plan Networks for Free Energy Calculations.","authors":"Benjamin Ries, Richard J Gowers, Hannah M Baumann, David W H Swenson, Michael M Henry, James R B Eastwood, Irfan Alibay, David Mobley","doi":"10.1021/acs.jcim.4c01710","DOIUrl":null,"url":null,"abstract":"<p><p>Alchemical free energy campaigns can be planned using graph theory by building networks that contain nodes representing molecules that are connected by possible transformations as edges. We introduce Konnektor, an open-source Python package, for systematically planning, modifying, and analyzing free energy calculation networks. Konnektor is designed to aid in the drug discovery process by enabling users to easily setup free energy campaigns using complex graph manipulation methods. The package contains functions for network operations including concatenation of networks, deletion of transformations, and clustering of molecules along with a framework for combining these tools with existing network generation algorithms to enable the development of more complex methods for network generation. A comparison of the various network layout features offered is carried out using toy data sets. Additionally, Konnektor contains visualization and analysis tools, making the investigation of network features much simpler. Besides the content of the package, the paper also offers application examples, demonstrating how Konnektor can be used and how the different networks perform from a graph theory perspective. Konnektor is freely available via GitHub at https://github.com/OpenFreeEnergy/konnektor under the permissive MIT License.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"8396-8403"},"PeriodicalIF":5.3000,"publicationDate":"2024-11-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11812579/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c01710","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/5 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
Alchemical free energy campaigns can be planned using graph theory by building networks that contain nodes representing molecules that are connected by possible transformations as edges. We introduce Konnektor, an open-source Python package, for systematically planning, modifying, and analyzing free energy calculation networks. Konnektor is designed to aid in the drug discovery process by enabling users to easily setup free energy campaigns using complex graph manipulation methods. The package contains functions for network operations including concatenation of networks, deletion of transformations, and clustering of molecules along with a framework for combining these tools with existing network generation algorithms to enable the development of more complex methods for network generation. A comparison of the various network layout features offered is carried out using toy data sets. Additionally, Konnektor contains visualization and analysis tools, making the investigation of network features much simpler. Besides the content of the package, the paper also offers application examples, demonstrating how Konnektor can be used and how the different networks perform from a graph theory perspective. Konnektor is freely available via GitHub at https://github.com/OpenFreeEnergy/konnektor under the permissive MIT License.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
