Estefanía Martínez-Barrios, Andrea Greco, José Cruzalegui, Sergi Cesar, Nuria Díez-Escuté, Patricia Cerralbo, Fredy Chipa, Irene Zschaeck, Leonel Slanovic, Alipio Mangas, Rocío Toro, Josep Brugada, Georgia Sarquella-Brugada, Oscar Campuzano
{"title":"Interpreting the actionable clinical role of rare variants associated with short QT syndrome.","authors":"Estefanía Martínez-Barrios, Andrea Greco, José Cruzalegui, Sergi Cesar, Nuria Díez-Escuté, Patricia Cerralbo, Fredy Chipa, Irene Zschaeck, Leonel Slanovic, Alipio Mangas, Rocío Toro, Josep Brugada, Georgia Sarquella-Brugada, Oscar Campuzano","doi":"10.1007/s00439-024-02713-x","DOIUrl":null,"url":null,"abstract":"<p><p>Genetic testing is recommended in the diagnosis of short QT syndrome. This rare inherited lethal entity is characterized by structural normal hearts with short QT intervals in the electrocardiogram. Few families diagnosed with this arrhythmogenic disease have been reported worldwide so far, impeding a comprehensive understanding of this syndrome. Unraveling the origin of the disease helps to the early identification of genetic carriers at risk. However, only rare variants with a definite deleterious role should be actionable in clinical practice. Our aim was to perform a comprehensive update and reinterpretation, according to the American College of Medical Genetics and Genomics recommendations of all rare variants currently associated with short QT syndrome. We identified 34 rare variants. Reanalysis showed that only nine variants played a deleterious role associated with a definite short QT syndrome phenotype. These variants were located in the four main genes: KCNQ1, KCNH2, KCNJ2 or SLC4A3. Additional rare variants located in other genes were associated with other conditions with phenotypic shortened QT intervals, but not definite diagnosis of short QT syndrome. Periodically updating of rare variants, especially those previously classified as unknown, helps to clarify the role of rare variants and translate genetic data into clinical practice.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"1499-1508"},"PeriodicalIF":3.6000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11576798/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-024-02713-x","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/6 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
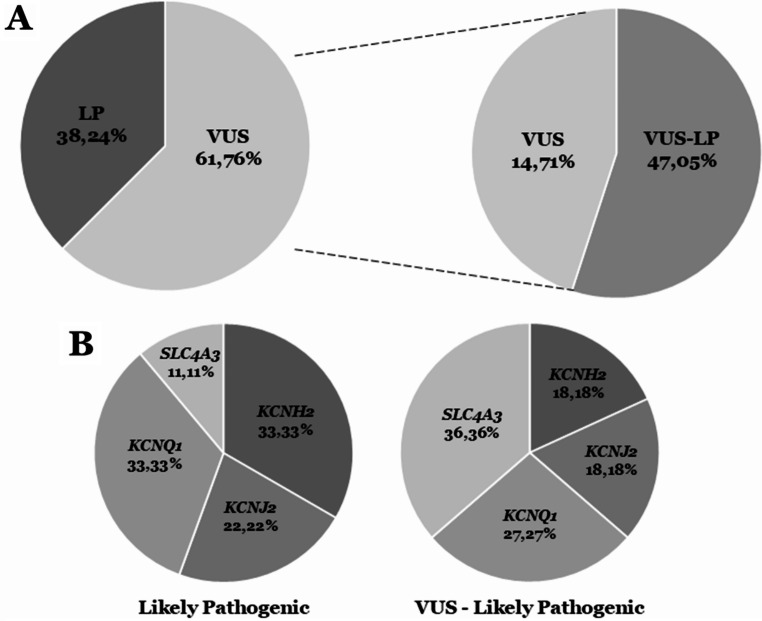
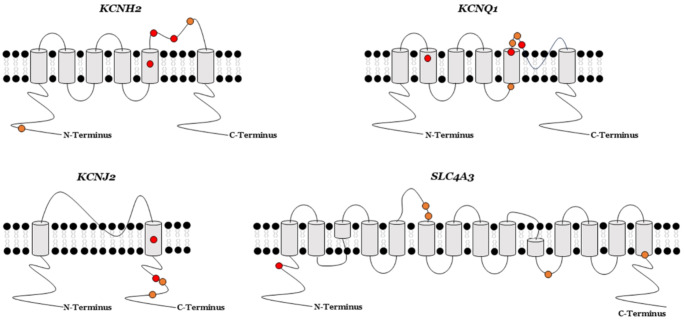
Genetic testing is recommended in the diagnosis of short QT syndrome. This rare inherited lethal entity is characterized by structural normal hearts with short QT intervals in the electrocardiogram. Few families diagnosed with this arrhythmogenic disease have been reported worldwide so far, impeding a comprehensive understanding of this syndrome. Unraveling the origin of the disease helps to the early identification of genetic carriers at risk. However, only rare variants with a definite deleterious role should be actionable in clinical practice. Our aim was to perform a comprehensive update and reinterpretation, according to the American College of Medical Genetics and Genomics recommendations of all rare variants currently associated with short QT syndrome. We identified 34 rare variants. Reanalysis showed that only nine variants played a deleterious role associated with a definite short QT syndrome phenotype. These variants were located in the four main genes: KCNQ1, KCNH2, KCNJ2 or SLC4A3. Additional rare variants located in other genes were associated with other conditions with phenotypic shortened QT intervals, but not definite diagnosis of short QT syndrome. Periodically updating of rare variants, especially those previously classified as unknown, helps to clarify the role of rare variants and translate genetic data into clinical practice.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
