Yile Chen, Kyoungyeul Lee, Junwoo Woo, Dong-Wook Kim, Changwon Keum, Giulia Babbi, Rita Casadio, Pier Luigi Martelli, Castrense Savojardo, Matteo Manfredi, Yang Shen, Yuanfei Sun, Panagiotis Katsonis, Olivier Lichtarge, Vikas Pejaver, David J Seward, Akash Kamandula, Constantina Bakolitsa, Steven E Brenner, Predrag Radivojac, Anne O'Donnell-Luria, Sean D Mooney, Shantanu Jain
{"title":"Evaluating predictors of kinase activity of STK11 variants identified in primary human non-small cell lung cancers.","authors":"Yile Chen, Kyoungyeul Lee, Junwoo Woo, Dong-Wook Kim, Changwon Keum, Giulia Babbi, Rita Casadio, Pier Luigi Martelli, Castrense Savojardo, Matteo Manfredi, Yang Shen, Yuanfei Sun, Panagiotis Katsonis, Olivier Lichtarge, Vikas Pejaver, David J Seward, Akash Kamandula, Constantina Bakolitsa, Steven E Brenner, Predrag Radivojac, Anne O'Donnell-Luria, Sean D Mooney, Shantanu Jain","doi":"10.1007/s00439-025-02726-0","DOIUrl":null,"url":null,"abstract":"<p><p>Critical evaluation of computational tools for predicting variant effects is important considering their increased use in disease diagnosis and driving molecular discoveries. In the sixth edition of the Critical Assessment of Genome Interpretation (CAGI) challenge, a dataset of 28 STK11 rare variants (27 missense, 1 single amino acid deletion), identified in primary non-small cell lung cancer biopsies, was experimentally assayed to characterize computational methods from four participating teams and five publicly available tools. Predictors demonstrated a high level of performance on key evaluation metrics, measuring correlation with the assay outputs and separating loss-of-function (LoF) variants from wildtype-like (WT-like) variants. The best participant model, 3Cnet, performed competitively with well-known tools. Unique to this challenge was that the functional data was generated with both biological and technical replicates, thus allowing the assessors to realistically establish maximum predictive performance based on experimental variability. Three out of the five publicly available tools and 3Cnet approached the performance of the assay replicates in separating LoF variants from WT-like variants. Surprisingly, REVEL, an often-used model, achieved a comparable correlation with the real-valued assay output as that seen for the experimental replicates. Performing variant interpretation by combining the new functional evidence with computational and population data evidence led to 16 new variants receiving a clinically actionable classification of likely pathogenic (LP) or likely benign (LB). Overall, the STK11 challenge highlights the utility of variant effect predictors in biomedical sciences and provides encouraging results for driving research in the field of computational genome interpretation.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"127-142"},"PeriodicalIF":3.6000,"publicationDate":"2025-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11976797/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-025-02726-0","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/12 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
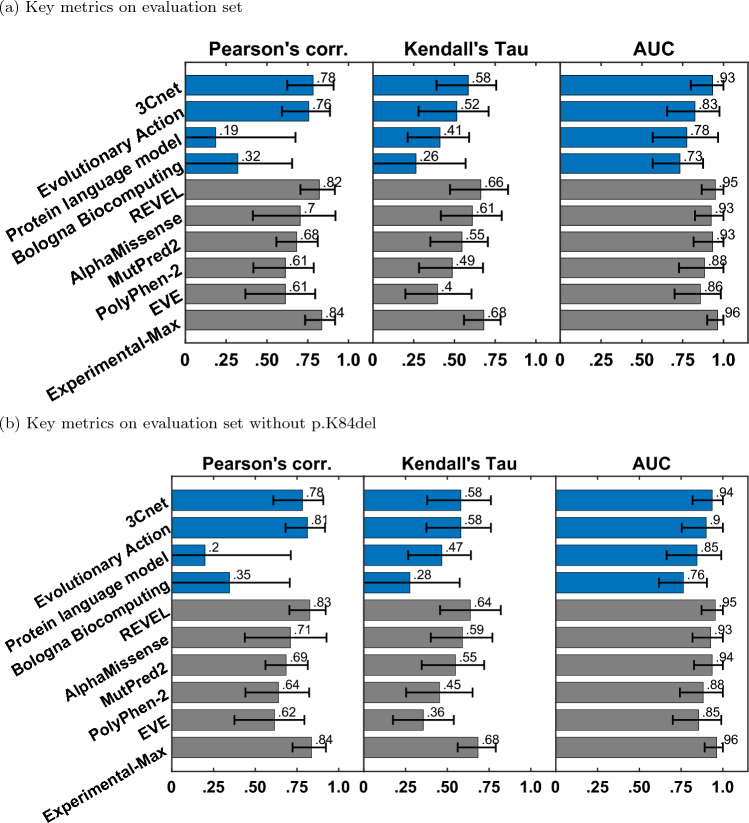
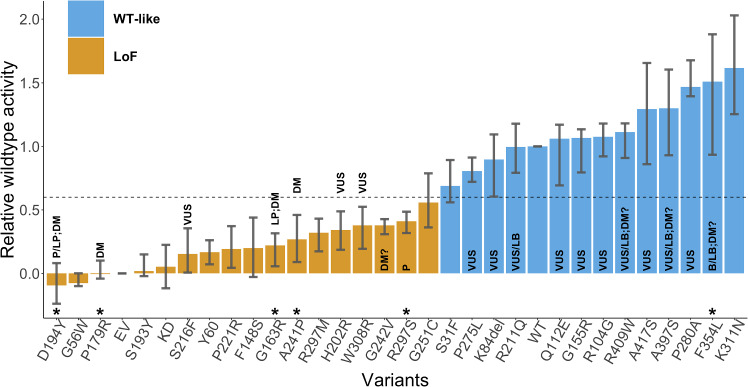
Critical evaluation of computational tools for predicting variant effects is important considering their increased use in disease diagnosis and driving molecular discoveries. In the sixth edition of the Critical Assessment of Genome Interpretation (CAGI) challenge, a dataset of 28 STK11 rare variants (27 missense, 1 single amino acid deletion), identified in primary non-small cell lung cancer biopsies, was experimentally assayed to characterize computational methods from four participating teams and five publicly available tools. Predictors demonstrated a high level of performance on key evaluation metrics, measuring correlation with the assay outputs and separating loss-of-function (LoF) variants from wildtype-like (WT-like) variants. The best participant model, 3Cnet, performed competitively with well-known tools. Unique to this challenge was that the functional data was generated with both biological and technical replicates, thus allowing the assessors to realistically establish maximum predictive performance based on experimental variability. Three out of the five publicly available tools and 3Cnet approached the performance of the assay replicates in separating LoF variants from WT-like variants. Surprisingly, REVEL, an often-used model, achieved a comparable correlation with the real-valued assay output as that seen for the experimental replicates. Performing variant interpretation by combining the new functional evidence with computational and population data evidence led to 16 new variants receiving a clinically actionable classification of likely pathogenic (LP) or likely benign (LB). Overall, the STK11 challenge highlights the utility of variant effect predictors in biomedical sciences and provides encouraging results for driving research in the field of computational genome interpretation.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
