Pei Song , Yuhang Zhou , Zishan Luo , Hang Zhang , Xi Sun , Sen Lu , Zepeng Jia , Hong Cui , Weizhi Tian , Rong Feng , Lingxia Jin , Hongkuan Yuan
{"title":"Valence electron matching law for MXene-based single-atom catalysts","authors":"Pei Song , Yuhang Zhou , Zishan Luo , Hang Zhang , Xi Sun , Sen Lu , Zepeng Jia , Hong Cui , Weizhi Tian , Rong Feng , Lingxia Jin , Hongkuan Yuan","doi":"10.1016/j.jechem.2024.10.006","DOIUrl":null,"url":null,"abstract":"<div><div>Single-atom catalysts (SACs) have attracted considerable interest in the fields of energy and environmental science due to their adjustable catalytic activity. In this study, we investigated the matching of valence electron numbers between single atoms and adsorbed intermediates (O, N, C, and H) in MXene-anchored SACs (M-Ti<sub>2</sub>C/M-Ti<sub>2</sub>CO<sub>2</sub>). The density functional theory results demonstrated that the sum of the valence electron number (<em>V</em><sub>M</sub>) of the interface-doped metal and the valence electron number (<em>V</em><sub>A</sub>) of the adsorbed intermediates in M-Ti<sub>2</sub>C followed the 10-valence electron matching law. Furthermore, based on the 10-valence electron matching law, we deduced that the sum of the valence electron number (<em>k</em>) and <em>V</em><sub>M</sub> for the molecular adsorption intermediate interactions in M-Ti<sub>2</sub>CO<sub>2</sub> adhered to the 11-valence electron matching law. Electrostatic repulsion between the interface electrons in M-Ti<sub>2</sub>CO<sub>2</sub> and H<sub>2</sub>O weakened the adsorption of intermediates. Furthermore, we applied the 11-valence electron matching law to guide the design of catalysts for nitrogen reduction reaction, specifically for N<sub>2</sub> → NNH conversion, in the M-Ti<sub>2</sub>CO<sub>2</sub> structure. The sure independence screening and sparsifying operator algorithm was used to fit a simple three-dimensional descriptor of the adsorbate (<em>R</em><sup>2</sup> up to 0.970) for catalyst design. Our study introduced a valence electron matching principle between doped metals (single atoms) and adsorbed intermediates (atomic and molecular) for MXene-based catalysts, providing new insights into the design of high-performance SACs.</div></div>","PeriodicalId":15728,"journal":{"name":"Journal of Energy Chemistry","volume":"101 ","pages":"Pages 641-650"},"PeriodicalIF":14.9000,"publicationDate":"2024-10-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Energy Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2095495624006995","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Energy","Score":null,"Total":0}
引用次数: 0
Abstract
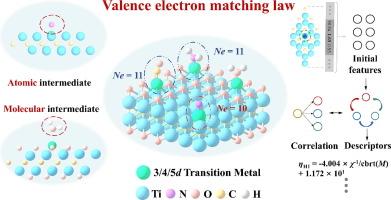
Single-atom catalysts (SACs) have attracted considerable interest in the fields of energy and environmental science due to their adjustable catalytic activity. In this study, we investigated the matching of valence electron numbers between single atoms and adsorbed intermediates (O, N, C, and H) in MXene-anchored SACs (M-Ti2C/M-Ti2CO2). The density functional theory results demonstrated that the sum of the valence electron number (VM) of the interface-doped metal and the valence electron number (VA) of the adsorbed intermediates in M-Ti2C followed the 10-valence electron matching law. Furthermore, based on the 10-valence electron matching law, we deduced that the sum of the valence electron number (k) and VM for the molecular adsorption intermediate interactions in M-Ti2CO2 adhered to the 11-valence electron matching law. Electrostatic repulsion between the interface electrons in M-Ti2CO2 and H2O weakened the adsorption of intermediates. Furthermore, we applied the 11-valence electron matching law to guide the design of catalysts for nitrogen reduction reaction, specifically for N2 → NNH conversion, in the M-Ti2CO2 structure. The sure independence screening and sparsifying operator algorithm was used to fit a simple three-dimensional descriptor of the adsorbate (R2 up to 0.970) for catalyst design. Our study introduced a valence electron matching principle between doped metals (single atoms) and adsorbed intermediates (atomic and molecular) for MXene-based catalysts, providing new insights into the design of high-performance SACs.
期刊介绍:
The Journal of Energy Chemistry, the official publication of Science Press and the Dalian Institute of Chemical Physics, Chinese Academy of Sciences, serves as a platform for reporting creative research and innovative applications in energy chemistry. It mainly reports on creative researches and innovative applications of chemical conversions of fossil energy, carbon dioxide, electrochemical energy and hydrogen energy, as well as the conversions of biomass and solar energy related with chemical issues to promote academic exchanges in the field of energy chemistry and to accelerate the exploration, research and development of energy science and technologies.
This journal focuses on original research papers covering various topics within energy chemistry worldwide, including:
Optimized utilization of fossil energy
Hydrogen energy
Conversion and storage of electrochemical energy
Capture, storage, and chemical conversion of carbon dioxide
Materials and nanotechnologies for energy conversion and storage
Chemistry in biomass conversion
Chemistry in the utilization of solar energy



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
