{"title":"Facilitated the discovery of new γ/γ′ Co-based superalloys by combining first-principles and machine learning","authors":"ZhaoJing Han, ShengBao Xia, ZeYu Chen, Yihui Guo, ZhaoXuan Li, Qinglian Huang, Xing-Jun Liu, Wei-Wei Xu","doi":"10.1038/s41524-024-01455-8","DOIUrl":null,"url":null,"abstract":"<p>Superalloys are indispensable materials for the fabrication of high-temperature components in aircraft engines. The discovery of a novel class of γ/γ′ Co-Al-W alloys has ignited a surge of interest in Co-based superalloys, with the aspiration to transcend the inherent constraints of their Ni-based counterparts. However, the conventional methodologies utilized in the design and advancement of new γ/γ′ Co-based superalloys are frequently characterized by their laborious and resource-intensive nature. In this study, we employed a coupled Density Functional Theory (DFT) and machine learning (ML) approach to predict and analyze the stability of the crucial γ′ phase, which is instrumental in expediting the discovery of γ/γ′ Co-based alloys. A dataset comprised of thousands of reliable formation (<i>H</i><sub>f</sub>) and decomposition (<i>H</i><sub>d</sub>) energies was obtained through high-throughput DFT calculations. Through regression model selection and feature engineering, our trained Random Forest (RF) model achieved prediction accuracies of 98.07% for <i>H</i><sub>f</sub> and 97.05% for <i>H</i><sub>d</sub>. Utilizing the well-trained RF model, we predicted the energies of over 150,000 ternary and quaternary γ′ phases within the Co-Ni-Fe-Cr-Al-W-Ti-Ta-V-Mo-Nb system. The energy analyses revealed that the presence of Ni, Nb, Ta, Ti, and V significantly reduced the <i>H</i><sub>f</sub> and the <i>H</i><sub>d</sub> of γ′, while Mo and W deteriorate the stability by increasing both energy values. Interestingly, although Al reduces the <i>H</i><sub>f</sub>, it increases <i>H</i><sub>d</sub>, thereby adversely affecting the stability of γ′. Applying domain-specific screening based on our knowledge, we identified 1049 out of >150,000 compositions likely to form stable γ′ phases, predominantly distributed across 11 Al-containing systems and 25 Al-free systems. Combining the analysis of CALPHAD method, we experimentally synthesized two new Co-based alloys with γ/γ′ dual-phase microstructures, corroborating the reliability of our theoretical prediction model.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"20 1","pages":""},"PeriodicalIF":9.4000,"publicationDate":"2024-11-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01455-8","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
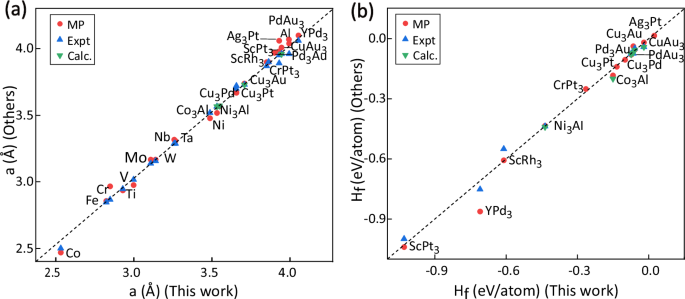
Superalloys are indispensable materials for the fabrication of high-temperature components in aircraft engines. The discovery of a novel class of γ/γ′ Co-Al-W alloys has ignited a surge of interest in Co-based superalloys, with the aspiration to transcend the inherent constraints of their Ni-based counterparts. However, the conventional methodologies utilized in the design and advancement of new γ/γ′ Co-based superalloys are frequently characterized by their laborious and resource-intensive nature. In this study, we employed a coupled Density Functional Theory (DFT) and machine learning (ML) approach to predict and analyze the stability of the crucial γ′ phase, which is instrumental in expediting the discovery of γ/γ′ Co-based alloys. A dataset comprised of thousands of reliable formation (Hf) and decomposition (Hd) energies was obtained through high-throughput DFT calculations. Through regression model selection and feature engineering, our trained Random Forest (RF) model achieved prediction accuracies of 98.07% for Hf and 97.05% for Hd. Utilizing the well-trained RF model, we predicted the energies of over 150,000 ternary and quaternary γ′ phases within the Co-Ni-Fe-Cr-Al-W-Ti-Ta-V-Mo-Nb system. The energy analyses revealed that the presence of Ni, Nb, Ta, Ti, and V significantly reduced the Hf and the Hd of γ′, while Mo and W deteriorate the stability by increasing both energy values. Interestingly, although Al reduces the Hf, it increases Hd, thereby adversely affecting the stability of γ′. Applying domain-specific screening based on our knowledge, we identified 1049 out of >150,000 compositions likely to form stable γ′ phases, predominantly distributed across 11 Al-containing systems and 25 Al-free systems. Combining the analysis of CALPHAD method, we experimentally synthesized two new Co-based alloys with γ/γ′ dual-phase microstructures, corroborating the reliability of our theoretical prediction model.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
