{"title":"Molecular electronic structure calculation via a quantum computer","authors":"Hamid Reza Naeij , Erfan Mahmoudi , Hossein Davoodi Yeganeh , Mohsen Akbari","doi":"10.1016/j.comptc.2024.114945","DOIUrl":null,"url":null,"abstract":"<div><div>Quantum computers can be used to calculate the electronic structure and estimate the ground state energy of many-electron molecular systems. In the present study, we implement the Variational Quantum Eigensolver (VQE) algorithm, as a hybrid quantum–classical algorithm to calculate the ground state energy of the molecules such as H<span><math><msubsup><mrow></mrow><mrow><mn>3</mn></mrow><mrow><mo>+</mo></mrow></msubsup></math></span>, OH<span><math><msup><mrow></mrow><mrow><mo>−</mo></mrow></msup></math></span>, HF and BH<span><math><msub><mrow></mrow><mrow><mn>3</mn></mrow></msub></math></span> in which the number of qubits has an increasing trend. We use the parity transformation for fermion to qubit encoding and the Unitary Coupled Cluster for Single and Double excitations (UCCSD) to construct an ansatz. We compare our quantum simulation results with the computational chemistry approaches including Full Configuration Interaction (FCI), as benchmark energy and Unrestricted Hartree–Fock (UHF), as a common computational method. Our results show that there is a good agreement between molecular ground state energy obtained from VQE and FCI. Moreover, the accuracy of the ground state energies obtained from VQE in our work is higher than the previously reported values. This work aims to benchmark the VQE algorithm to calculate the electronic ground state energy for a new set of molecules that can be good candidates for molecular simulation on a real quantum computer.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1242 ","pages":"Article 114945"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004845","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/11/4 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
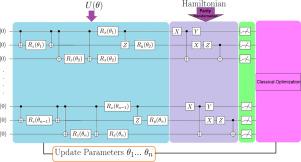
Quantum computers can be used to calculate the electronic structure and estimate the ground state energy of many-electron molecular systems. In the present study, we implement the Variational Quantum Eigensolver (VQE) algorithm, as a hybrid quantum–classical algorithm to calculate the ground state energy of the molecules such as H, OH, HF and BH in which the number of qubits has an increasing trend. We use the parity transformation for fermion to qubit encoding and the Unitary Coupled Cluster for Single and Double excitations (UCCSD) to construct an ansatz. We compare our quantum simulation results with the computational chemistry approaches including Full Configuration Interaction (FCI), as benchmark energy and Unrestricted Hartree–Fock (UHF), as a common computational method. Our results show that there is a good agreement between molecular ground state energy obtained from VQE and FCI. Moreover, the accuracy of the ground state energies obtained from VQE in our work is higher than the previously reported values. This work aims to benchmark the VQE algorithm to calculate the electronic ground state energy for a new set of molecules that can be good candidates for molecular simulation on a real quantum computer.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
