Giorgia Mandrile, Barbara Cellini, Pietro Manuel Ferraro
{"title":"Effect of the allelic background on the phenotype of primary hyperoxaluria type I.","authors":"Giorgia Mandrile, Barbara Cellini, Pietro Manuel Ferraro","doi":"10.1097/MNH.0000000000001057","DOIUrl":null,"url":null,"abstract":"<p><strong>Purpose of review: </strong>Primary hyperoxaluria type 1 (PH1) is an autosomal recessive disorder of hepatic glyoxylate metabolism leading to nephrolithiasis and kidney failure. PH1 is caused by mutations on the AGXT gene encoding alanine:glyoxylate aminotransferase (AGT). The AGXT gene has two haplotypes, the major (Ma) and the minor (mi) alleles. This review summarizes the role of the minor allele on the molecular pathogenesis and the clinical manifestations of PH1.</p><p><strong>Recent findings: </strong>PH1 shows high genetic variability and significant interindividual variability. Although the minor haplotype is not pathogenic on its own, it may be crucial for the pathogenicity of some mutations or amplify the effect of others, thus affecting both symptoms and responsiveness to Vitamin B6, the only pharmacological treatment effective in a selected group of PH1 patients.</p><p><strong>Summary: </strong>In the last years, new drugs based on RNA-interference are available for patients nonresponsive to Vitamin B6, but no specific biomarkers are available to predict disease course and severity. Therefore, a clinical assessment of PH1 taking into account molecular analysis of the mutations and the allelic background and the possible synergism among polymorphic and pathogenic variants should be encouraged to promote approaches of personalized medicine that improve the management of available resources.</p>","PeriodicalId":10960,"journal":{"name":"Current Opinion in Nephrology and Hypertension","volume":" ","pages":"177-183"},"PeriodicalIF":2.4000,"publicationDate":"2025-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11789592/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current Opinion in Nephrology and Hypertension","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1097/MNH.0000000000001057","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/12/6 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"PERIPHERAL VASCULAR DISEASE","Score":null,"Total":0}
引用次数: 0
Abstract
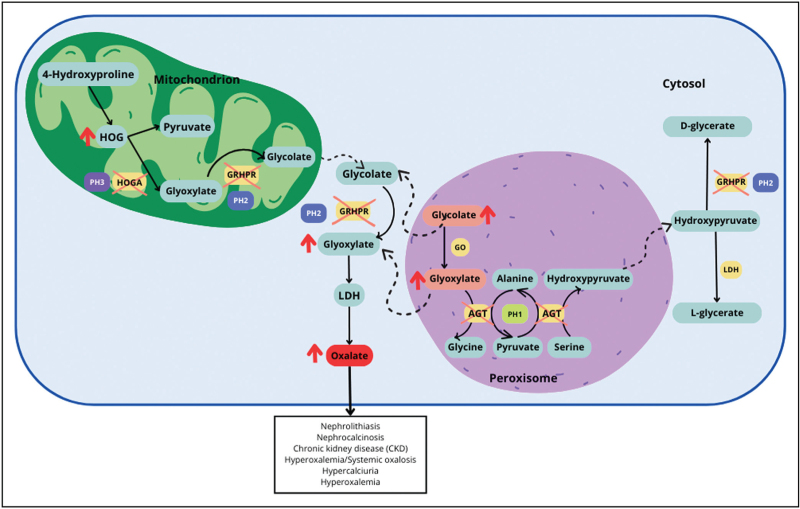
Purpose of review: Primary hyperoxaluria type 1 (PH1) is an autosomal recessive disorder of hepatic glyoxylate metabolism leading to nephrolithiasis and kidney failure. PH1 is caused by mutations on the AGXT gene encoding alanine:glyoxylate aminotransferase (AGT). The AGXT gene has two haplotypes, the major (Ma) and the minor (mi) alleles. This review summarizes the role of the minor allele on the molecular pathogenesis and the clinical manifestations of PH1.
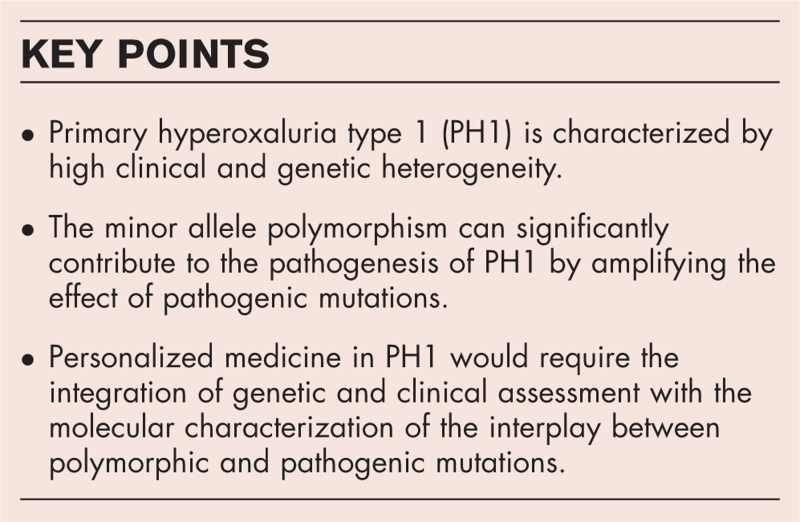
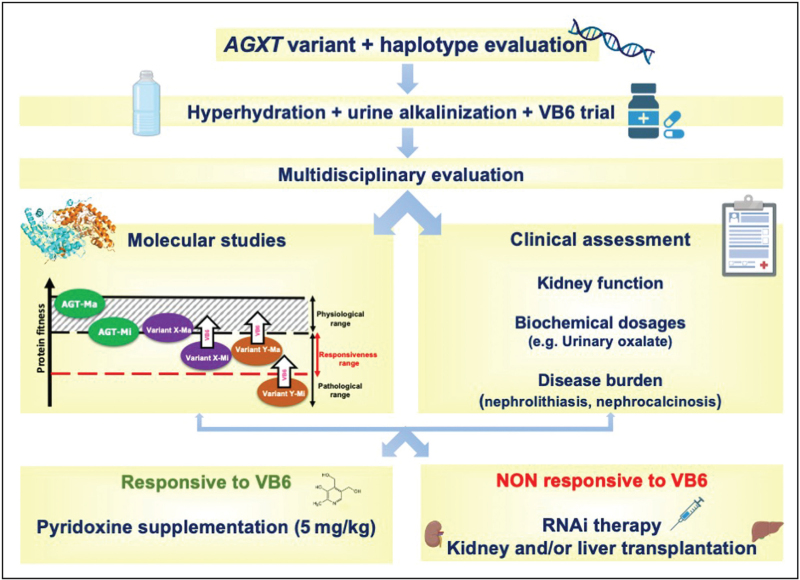
Recent findings: PH1 shows high genetic variability and significant interindividual variability. Although the minor haplotype is not pathogenic on its own, it may be crucial for the pathogenicity of some mutations or amplify the effect of others, thus affecting both symptoms and responsiveness to Vitamin B6, the only pharmacological treatment effective in a selected group of PH1 patients.
Summary: In the last years, new drugs based on RNA-interference are available for patients nonresponsive to Vitamin B6, but no specific biomarkers are available to predict disease course and severity. Therefore, a clinical assessment of PH1 taking into account molecular analysis of the mutations and the allelic background and the possible synergism among polymorphic and pathogenic variants should be encouraged to promote approaches of personalized medicine that improve the management of available resources.
期刊介绍:
A reader-friendly resource, Current Opinion in Nephrology and Hypertension provides an up-to-date account of the most important advances in the field of nephrology and hypertension. Each issue contains either two or three sections delivering a diverse and comprehensive coverage of all the key issues, including pathophysiology of hypertension, circulation and hemodynamics, and clinical nephrology. Current Opinion in Nephrology and Hypertension is an indispensable journal for the busy clinician, researcher or student.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
