Hongwei Du, Jiamin Wang, Jian Hui, Lanting Zhang, Hong Wang
{"title":"DenseGNN: universal and scalable deeper graph neural networks for high-performance property prediction in crystals and molecules","authors":"Hongwei Du, Jiamin Wang, Jian Hui, Lanting Zhang, Hong Wang","doi":"10.1038/s41524-024-01444-x","DOIUrl":null,"url":null,"abstract":"<p>Modern generative models based on deep learning have made it possible to design millions of hypothetical materials. To screen these candidate materials and identify promising new materials, we need fast and accurate models to predict material properties. Graphical neural networks (GNNs) have become a current research focus due to their ability to directly act on the graphical representation of molecules and materials, enabling comprehensive capture of important information and showing excellent performance in predicting material properties. Nevertheless, GNNs still face several key problems in practical applications: First, although existing nested graph network strategies increase critical structural information such as bond angles, they significantly increase the number of trainable parameters in the model, resulting in a increase in training costs; Second, extending GNN models to broader domains such as molecules, crystalline materials, and catalysis, as well as adapting to small data sets, remains a challenge. Finally, the scalability of GNN models is limited by the over-smoothing problem. To address these issues, we propose the DenseGNN model, which combines Dense Connectivity Network (DCN), hierarchical node-edge-graph residual networks (HRN), and Local Structure Order Parameters Embedding (LOPE) strategies to create a universal, scalable, and efficient GNN model. We have achieved state-of-the-art performance (SOAT) on several datasets, including JARVIS-DFT, Materials Project, QM9, Lipop, FreeSolv, ESOL, and OC22, demonstrating the generality and scalability of our approach. By merging DCN and LOPE strategies into GNN models in computing, crystal materials, and molecules, we have improved the performance of models such as GIN, Schnet, and Hamnet on materials datasets such as Matbench. The LOPE strategy optimizes the embedding representation of atoms and allows our model to train efficiently with a minimal level of edge connections. This substantially reduces computational costs and shortens the time required to train large GNNs while maintaining accuracy. Our technique not only supports building deeper GNNs and avoids performance penalties experienced by other models, but is also applicable to a variety of applications that require large deep learning models. Furthermore, our study demonstrates that by using structural embeddings from pre-trained models, our model not only outperforms other GNNs in distinguishing crystal structures but also approaches the standard X-ray diffraction (XRD) method.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"27 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2024-12-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01444-x","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
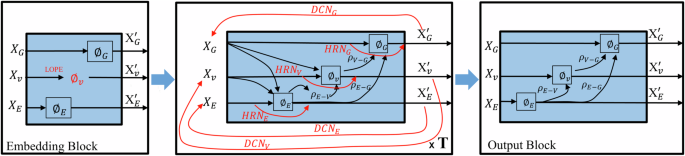
Modern generative models based on deep learning have made it possible to design millions of hypothetical materials. To screen these candidate materials and identify promising new materials, we need fast and accurate models to predict material properties. Graphical neural networks (GNNs) have become a current research focus due to their ability to directly act on the graphical representation of molecules and materials, enabling comprehensive capture of important information and showing excellent performance in predicting material properties. Nevertheless, GNNs still face several key problems in practical applications: First, although existing nested graph network strategies increase critical structural information such as bond angles, they significantly increase the number of trainable parameters in the model, resulting in a increase in training costs; Second, extending GNN models to broader domains such as molecules, crystalline materials, and catalysis, as well as adapting to small data sets, remains a challenge. Finally, the scalability of GNN models is limited by the over-smoothing problem. To address these issues, we propose the DenseGNN model, which combines Dense Connectivity Network (DCN), hierarchical node-edge-graph residual networks (HRN), and Local Structure Order Parameters Embedding (LOPE) strategies to create a universal, scalable, and efficient GNN model. We have achieved state-of-the-art performance (SOAT) on several datasets, including JARVIS-DFT, Materials Project, QM9, Lipop, FreeSolv, ESOL, and OC22, demonstrating the generality and scalability of our approach. By merging DCN and LOPE strategies into GNN models in computing, crystal materials, and molecules, we have improved the performance of models such as GIN, Schnet, and Hamnet on materials datasets such as Matbench. The LOPE strategy optimizes the embedding representation of atoms and allows our model to train efficiently with a minimal level of edge connections. This substantially reduces computational costs and shortens the time required to train large GNNs while maintaining accuracy. Our technique not only supports building deeper GNNs and avoids performance penalties experienced by other models, but is also applicable to a variety of applications that require large deep learning models. Furthermore, our study demonstrates that by using structural embeddings from pre-trained models, our model not only outperforms other GNNs in distinguishing crystal structures but also approaches the standard X-ray diffraction (XRD) method.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
