{"title":"DFAST_QC: quality assessment and taxonomic identification tool for prokaryotic Genomes.","authors":"Mohamed Elmanzalawi, Takatomo Fujisawa, Hiroshi Mori, Yasukazu Nakamura, Yasuhiro Tanizawa","doi":"10.1186/s12859-024-06030-y","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Accurate taxonomic classification in genome databases is essential for reliable biological research and effective data sharing. Mislabeling or inaccuracies in genome annotations can lead to incorrect scientific conclusions and hinder the reproducibility of research findings. Despite advances in genome analysis techniques, challenges persist in ensuring precise and reliable taxonomic assignments. Existing tools for genome verification often involve extensive computational resources or lengthy processing times, which can limit their accessibility and scalability for large-scale projects. There is a need for more efficient, user-friendly solutions that can handle diverse datasets and provide accurate results with minimal computational demands. This work aimed to address these challenges by introducing a novel tool that enhances taxonomic accuracy, offers a user-friendly interface, and supports large-scale analyses.</p><p><strong>Results: </strong>We introduce a novel tool for the quality control and taxonomic classification tool of prokaryotic genomes, called DFAST_QC, which is available as both a command-line tool and a web service. DFAST_QC can quickly identify species based on NCBI and GTDB taxonomies by combining genome-distance calculations using MASH with ANI calculations using Skani. We evaluated DFAST_QC's performance in species identification and found it to be highly consistent with existing taxonomic standards, successfully identifying species across diverse datasets. In several cases, DFAST_QC identified potential mislabeling of species names in public databases and highlighted discrepancies in current classifications, demonstrating its capability to uncover errors and enhance taxonomic accuracy. Additionally, the tool's efficient design allows it to operate smoothly on local machines with minimal computational requirements, making it a practical choice for large-scale genome projects.</p><p><strong>Conclusions: </strong>DFAST_QC is a reliable and efficient tool for accurate taxonomic identification and genome quality control, well-suited for large-scale genomic studies. Its compatibility with limited-resource environments, combined with its user-friendly design, ensures seamless integration into existing workflows. DFAST_QC's ability to refine species assignments in public databases highlights its value as a complementary tool for maintaining and enhancing the accuracy of taxonomic data in genomic research. The web version is available at https://dfast.ddbj.nig.ac.jp/dqc/submit/ , and the source code for local use can be found at https://github.com/nigyta/dfast_qc .</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"3"},"PeriodicalIF":3.3000,"publicationDate":"2025-01-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11705978/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-024-06030-y","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Accurate taxonomic classification in genome databases is essential for reliable biological research and effective data sharing. Mislabeling or inaccuracies in genome annotations can lead to incorrect scientific conclusions and hinder the reproducibility of research findings. Despite advances in genome analysis techniques, challenges persist in ensuring precise and reliable taxonomic assignments. Existing tools for genome verification often involve extensive computational resources or lengthy processing times, which can limit their accessibility and scalability for large-scale projects. There is a need for more efficient, user-friendly solutions that can handle diverse datasets and provide accurate results with minimal computational demands. This work aimed to address these challenges by introducing a novel tool that enhances taxonomic accuracy, offers a user-friendly interface, and supports large-scale analyses.
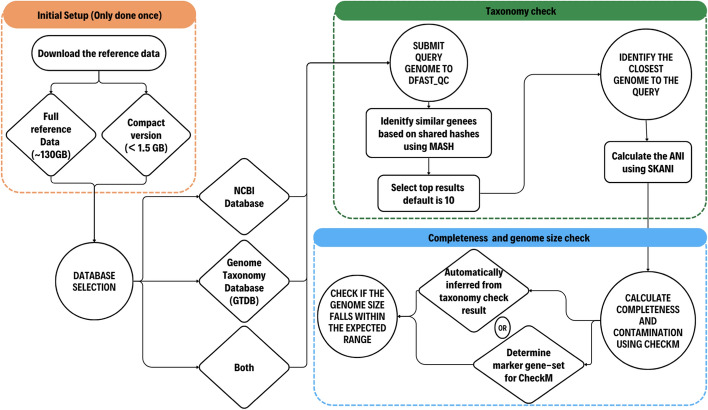
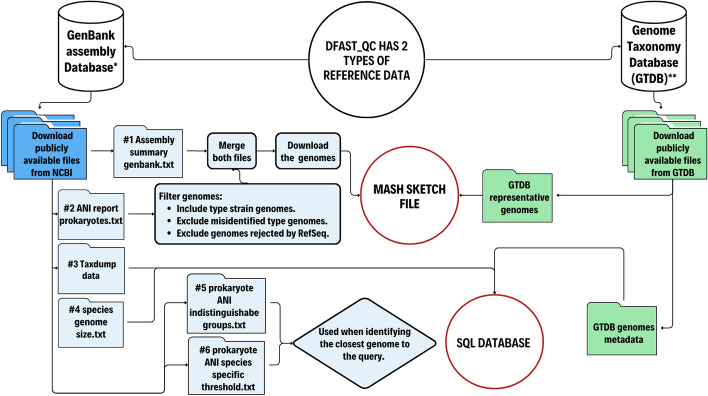
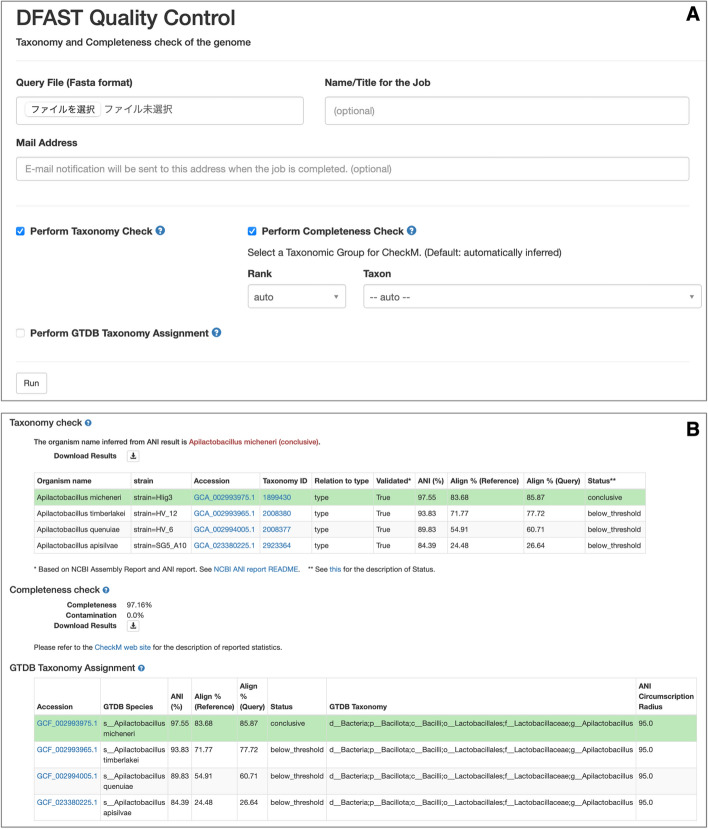
Results: We introduce a novel tool for the quality control and taxonomic classification tool of prokaryotic genomes, called DFAST_QC, which is available as both a command-line tool and a web service. DFAST_QC can quickly identify species based on NCBI and GTDB taxonomies by combining genome-distance calculations using MASH with ANI calculations using Skani. We evaluated DFAST_QC's performance in species identification and found it to be highly consistent with existing taxonomic standards, successfully identifying species across diverse datasets. In several cases, DFAST_QC identified potential mislabeling of species names in public databases and highlighted discrepancies in current classifications, demonstrating its capability to uncover errors and enhance taxonomic accuracy. Additionally, the tool's efficient design allows it to operate smoothly on local machines with minimal computational requirements, making it a practical choice for large-scale genome projects.
Conclusions: DFAST_QC is a reliable and efficient tool for accurate taxonomic identification and genome quality control, well-suited for large-scale genomic studies. Its compatibility with limited-resource environments, combined with its user-friendly design, ensures seamless integration into existing workflows. DFAST_QC's ability to refine species assignments in public databases highlights its value as a complementary tool for maintaining and enhancing the accuracy of taxonomic data in genomic research. The web version is available at https://dfast.ddbj.nig.ac.jp/dqc/submit/ , and the source code for local use can be found at https://github.com/nigyta/dfast_qc .
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
