Diagnostic delay in cerebral creatine deficiency disorders: lessons learned from a cross-sectional single center study, and guanidinoacetate and creatine measurements in Switzerland between 2015 and 2023.
Christina Kaufman, Anaïs D'Andrea, Annette Hackenberg, Martin Poms, Olivier Braissant, Johannes Häberle
{"title":"Diagnostic delay in cerebral creatine deficiency disorders: lessons learned from a cross-sectional single center study, and guanidinoacetate and creatine measurements in Switzerland between 2015 and 2023.","authors":"Christina Kaufman, Anaïs D'Andrea, Annette Hackenberg, Martin Poms, Olivier Braissant, Johannes Häberle","doi":"10.1186/s40348-024-00188-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Cerebral creatine deficiency disorders (CCDD) are rare diseases caused by defects in the enzymes L-arginine: glycine amidinotransferase (AGAT) or guanidinoacetate-N-methyltransferase (GAMT), which are involved in synthesis of creatine; or by a defect in the creatine transporter (CRTR), which is essential for uptake of creatine as important energy source into the target cells. Patients with CCDD can present with a variety of unspecific symptoms: global developmental delay, speech-language disorder, behavioral abnormalities and seizures. Early treatment initiation is essential in AGAT and GAMT deficiencies to achieve a favorable outcome. This study describes the CCDD patient cohort in a single center, and an analysis of the referrals to two Swiss laboratories in Lausanne and Zurich between 2015 and 2023 for the two marker metabolites guanidinoacetate and creatine.</p><p><strong>Results: </strong>The patient cohort comprised 6 patients (defects: 2 GAMT, 4 CRTR), who were initially seen by different subspecialties depending on first symptoms. There was a diagnostic and therapeutic delay between 3 and 32 months (mean 13.8). Numbers of biomarker requests showed a constant increase during the study period, with a majority of tests performed in urine, the preferred sample for CCDD detection. Almost all samples (93.3%) were sent in by large hospitals (mainly from neurology, developmental pediatrics and metabolism) and only few (5.2%) by pediatricians in private practice, although those usually see the patients first.</p><p><strong>Conclusions: </strong>The data from this study demonstrate a relevant delay in identifying patients with these rare conditions, and a predominance of biomarker analysis requested from pediatric subspecialties that are involved in patient management often long after occurrence of symptoms. To reduce the diagnostic delay and the outcome of patients, the current practice of sample referral should be reflected and first-contact healthcare providers should be encouraged to initiate selective screening.</p>","PeriodicalId":74215,"journal":{"name":"Molecular and cellular pediatrics","volume":"12 1","pages":"1"},"PeriodicalIF":3.4000,"publicationDate":"2025-01-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11751272/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular and cellular pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40348-024-00188-4","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 0
Abstract
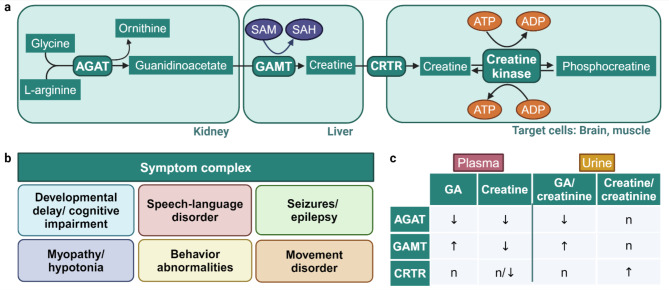
Background: Cerebral creatine deficiency disorders (CCDD) are rare diseases caused by defects in the enzymes L-arginine: glycine amidinotransferase (AGAT) or guanidinoacetate-N-methyltransferase (GAMT), which are involved in synthesis of creatine; or by a defect in the creatine transporter (CRTR), which is essential for uptake of creatine as important energy source into the target cells. Patients with CCDD can present with a variety of unspecific symptoms: global developmental delay, speech-language disorder, behavioral abnormalities and seizures. Early treatment initiation is essential in AGAT and GAMT deficiencies to achieve a favorable outcome. This study describes the CCDD patient cohort in a single center, and an analysis of the referrals to two Swiss laboratories in Lausanne and Zurich between 2015 and 2023 for the two marker metabolites guanidinoacetate and creatine.
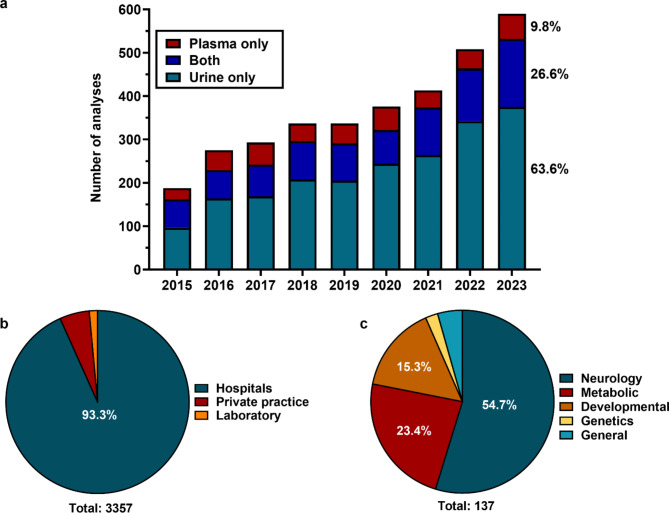
Results: The patient cohort comprised 6 patients (defects: 2 GAMT, 4 CRTR), who were initially seen by different subspecialties depending on first symptoms. There was a diagnostic and therapeutic delay between 3 and 32 months (mean 13.8). Numbers of biomarker requests showed a constant increase during the study period, with a majority of tests performed in urine, the preferred sample for CCDD detection. Almost all samples (93.3%) were sent in by large hospitals (mainly from neurology, developmental pediatrics and metabolism) and only few (5.2%) by pediatricians in private practice, although those usually see the patients first.
Conclusions: The data from this study demonstrate a relevant delay in identifying patients with these rare conditions, and a predominance of biomarker analysis requested from pediatric subspecialties that are involved in patient management often long after occurrence of symptoms. To reduce the diagnostic delay and the outcome of patients, the current practice of sample referral should be reflected and first-contact healthcare providers should be encouraged to initiate selective screening.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
