{"title":"Crystal Structure Prediction of Cs–Te with Supervised Machine Learning","authors":"Holger-Dietrich Saßnick, Caterina Cocchi","doi":"10.1002/adts.202401344","DOIUrl":null,"url":null,"abstract":"<p>Crystal structure prediction methods aim to determine the ground-state crystal structure for a given material. The vast combinatorial space associated with this problem makes conventional methods computationally prohibitive for routine use. To overcome these limitations, a novel approach combining high-throughput density functional theory calculations with machine learning is proposed. It predicts stable crystal structures within binary and ternary systems by systematically evaluating various structural descriptors and machine learning algorithms. The superiority of models based on atomic coordination environments is shown, with transfer-learned graph neural networks emerging as a particularly promising technique. By validating the proposed method on Cs–Te crystals, its ability to generate stable crystal structures is proved, suggesting its potential for advancing established computational schemes.</p>","PeriodicalId":7219,"journal":{"name":"Advanced Theory and Simulations","volume":"8 5","pages":""},"PeriodicalIF":2.9000,"publicationDate":"2025-01-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/adts.202401344","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Advanced Theory and Simulations","FirstCategoryId":"5","ListUrlMain":"https://advanced.onlinelibrary.wiley.com/doi/10.1002/adts.202401344","RegionNum":4,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
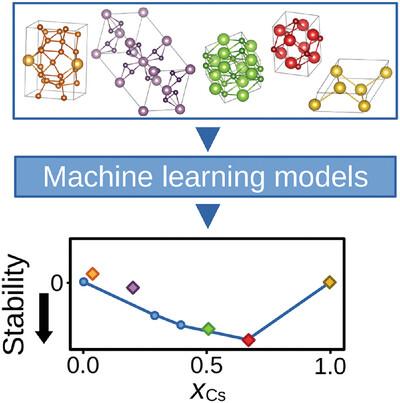
Crystal structure prediction methods aim to determine the ground-state crystal structure for a given material. The vast combinatorial space associated with this problem makes conventional methods computationally prohibitive for routine use. To overcome these limitations, a novel approach combining high-throughput density functional theory calculations with machine learning is proposed. It predicts stable crystal structures within binary and ternary systems by systematically evaluating various structural descriptors and machine learning algorithms. The superiority of models based on atomic coordination environments is shown, with transfer-learned graph neural networks emerging as a particularly promising technique. By validating the proposed method on Cs–Te crystals, its ability to generate stable crystal structures is proved, suggesting its potential for advancing established computational schemes.
期刊介绍:
Advanced Theory and Simulations is an interdisciplinary, international, English-language journal that publishes high-quality scientific results focusing on the development and application of theoretical methods, modeling and simulation approaches in all natural science and medicine areas, including:
materials, chemistry, condensed matter physics
engineering, energy
life science, biology, medicine
atmospheric/environmental science, climate science
planetary science, astronomy, cosmology
method development, numerical methods, statistics







 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
