Weina Zhao, Chang Shen, Anil Kumar Tummanapelli, Ming Wah Wong
{"title":"Computational Insights Into Corrosion Inhibition Mechanism: Dissociation of Imidazole on Iron Surface","authors":"Weina Zhao, Chang Shen, Anil Kumar Tummanapelli, Ming Wah Wong","doi":"10.1002/jcc.70047","DOIUrl":null,"url":null,"abstract":"<p>Corrosion inhibitors are widely used to mitigate safety risks and economic losses in engineering, yet post-adsorption processes remain underexplored. In this study, we employed density functional theory calculations with a periodic model to investigate the dissociation mechanisms of imidazole on the Fe(100) surface. Imidazole was found to adsorb optimally in a parallel orientation, with an adsorption energy of −0.88 eV. We explored two dissociation pathways: C<span></span>H and N<span></span>H bond cleavages and found C<span></span>H dissociation having a lower activation barrier of 0.46 eV. Intriguingly, an alternative indirect route C<span></span>H dissociation pathway involving a tilted intermediate state was found to be competitive. Both indirect and direct C<span></span>H dissociation pathways are energetically more favorable than N<span></span>H cleavage. Molecular dynamics simulations reveal that indirect C<span></span>H dissociation occurs rapidly. This study proposes an alternative protective mechanism involving dissociated imidazole inhibitors, offering new insights for corrosion inhibitor design.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 3","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-01-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.70047","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70047","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
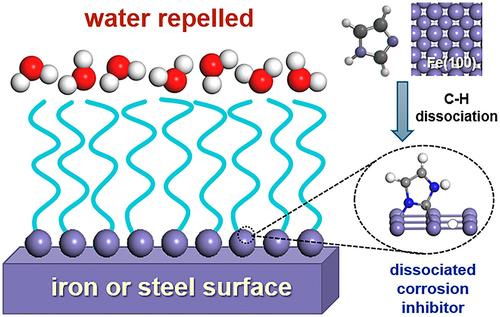
Corrosion inhibitors are widely used to mitigate safety risks and economic losses in engineering, yet post-adsorption processes remain underexplored. In this study, we employed density functional theory calculations with a periodic model to investigate the dissociation mechanisms of imidazole on the Fe(100) surface. Imidazole was found to adsorb optimally in a parallel orientation, with an adsorption energy of −0.88 eV. We explored two dissociation pathways: CH and NH bond cleavages and found CH dissociation having a lower activation barrier of 0.46 eV. Intriguingly, an alternative indirect route CH dissociation pathway involving a tilted intermediate state was found to be competitive. Both indirect and direct CH dissociation pathways are energetically more favorable than NH cleavage. Molecular dynamics simulations reveal that indirect CH dissociation occurs rapidly. This study proposes an alternative protective mechanism involving dissociated imidazole inhibitors, offering new insights for corrosion inhibitor design.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
