Gia Linh Hoang, Manuel Röck, Aldo Tancredi, Thomas Magauer, Davide Mandelli, Jörg B. Schulz, Sybille Krauss, Giulia Rossetti, Martin Tollinger, Paolo Carloni
{"title":"Refining Ligand Poses in RNA/Ligand Complexes of Pharmaceutical Relevance: A Perspective by QM/MM Simulations and NMR Measurements","authors":"Gia Linh Hoang, Manuel Röck, Aldo Tancredi, Thomas Magauer, Davide Mandelli, Jörg B. Schulz, Sybille Krauss, Giulia Rossetti, Martin Tollinger, Paolo Carloni","doi":"10.1021/acs.jpclett.4c03456","DOIUrl":null,"url":null,"abstract":"Predicting the binding poses of ligands targeting RNAs is challenging. Here, we propose that using first-principles quantum mechanics/molecular mechanics (QM/MM) simulations, which incorporate automatically polarization effects, can help refine the structural determinants of ligand/RNA complexes in aqueous solution. In fact, recent advances in massively parallel computer architectures (such as exascale machines), combined with the power of machine learning, are greatly expanding the domain of applicability of these types of notoriously expensive simulations. We corroborate this proposal by carrying out a QM/MM-based study on a ligand targeting CAG repeat-RNA, involved in Huntington’s disease. The calculations indeed show a clear improvement in the ligand binding properties, and they are consistent with the NMR measurements, also performed here. Thus, this type of approach may be useful for practical applications in the design of ligands targeting RNA in the near future.","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"62 1","pages":""},"PeriodicalIF":4.6000,"publicationDate":"2025-02-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpclett.4c03456","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
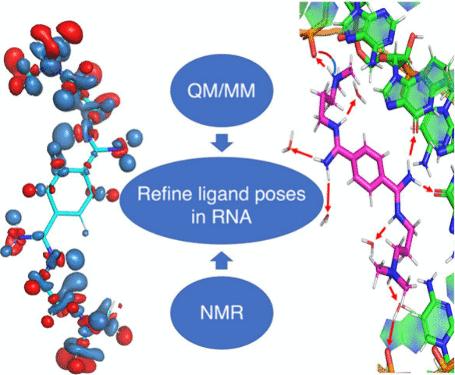
Predicting the binding poses of ligands targeting RNAs is challenging. Here, we propose that using first-principles quantum mechanics/molecular mechanics (QM/MM) simulations, which incorporate automatically polarization effects, can help refine the structural determinants of ligand/RNA complexes in aqueous solution. In fact, recent advances in massively parallel computer architectures (such as exascale machines), combined with the power of machine learning, are greatly expanding the domain of applicability of these types of notoriously expensive simulations. We corroborate this proposal by carrying out a QM/MM-based study on a ligand targeting CAG repeat-RNA, involved in Huntington’s disease. The calculations indeed show a clear improvement in the ligand binding properties, and they are consistent with the NMR measurements, also performed here. Thus, this type of approach may be useful for practical applications in the design of ligands targeting RNA in the near future.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
