Abigail P. Onufer , Joshua Chang Mell , Laura Cort , Abhishek Rao , Nontokozo V. Mdluli , Alison J. Carey
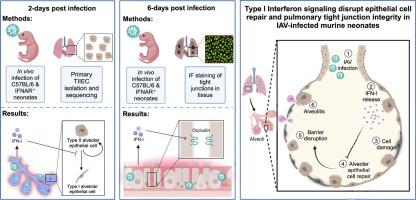
{"title":"Influenza virus-induced type I interferons disrupt alveolar epithelial repair and tight junction integrity in the developing lung","authors":"Abigail P. Onufer , Joshua Chang Mell , Laura Cort , Abhishek Rao , Nontokozo V. Mdluli , Alison J. Carey","doi":"10.1016/j.mucimm.2025.02.002","DOIUrl":null,"url":null,"abstract":"<div><div>Recently, we demonstrated that influenza A virus (IAV)-infected murine neonates lacking a functional IFN-I receptor (IFNAR<sup>−/−</sup>) had significantly improved survival and reduced lung pathology relative to wild-type (WT) neonates. In direct contrast, adult IFNAR<sup>−/−</sup> mice display enhanced morbidity following IAV infection relative to WT adults. We hypothesized that IAV-induced IFN-I signaling in primary neonatal type II alveolar epithelial cells (TIIECs), the main cell type of IAV infection and initiator of host response in the lung, contributed to age-specific viral pathogenesis. Multifactorial transcriptional analysis of purified TIIECs revealed age, not infection status, as the primary driver of transcriptional differences in TIIECs. Subsequent pathway analysis demonstrated IAV-infected IFNAR<sup>−/−</sup> neonates significantly upregulated cell proliferation, tissue repair and tight junction genes at 2-days post-infection (dpi), compared to WT neonates. Next, to determine if these growth and repair differences persisted later in infection, targeted analysis of repair gene expression and immunofluorescent quantification of pulmonary sealing tight junction molecules ZO-1 and occludin was performed at 6-dpi. Relative to WT neonates, IFNAR<sup>−/−</sup> neonates had significantly higher whole lung occludin staining and repair gene expression. Together, our data demonstrates IFN-I signaling is extremely pathogenic in the developing lung by disrupting alveolar repair and pulmonary barrier integrity.</div></div>","PeriodicalId":18877,"journal":{"name":"Mucosal Immunology","volume":"18 3","pages":"Pages 607-619"},"PeriodicalIF":7.6000,"publicationDate":"2025-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Mucosal Immunology","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1933021925000200","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Recently, we demonstrated that influenza A virus (IAV)-infected murine neonates lacking a functional IFN-I receptor (IFNAR−/−) had significantly improved survival and reduced lung pathology relative to wild-type (WT) neonates. In direct contrast, adult IFNAR−/− mice display enhanced morbidity following IAV infection relative to WT adults. We hypothesized that IAV-induced IFN-I signaling in primary neonatal type II alveolar epithelial cells (TIIECs), the main cell type of IAV infection and initiator of host response in the lung, contributed to age-specific viral pathogenesis. Multifactorial transcriptional analysis of purified TIIECs revealed age, not infection status, as the primary driver of transcriptional differences in TIIECs. Subsequent pathway analysis demonstrated IAV-infected IFNAR−/− neonates significantly upregulated cell proliferation, tissue repair and tight junction genes at 2-days post-infection (dpi), compared to WT neonates. Next, to determine if these growth and repair differences persisted later in infection, targeted analysis of repair gene expression and immunofluorescent quantification of pulmonary sealing tight junction molecules ZO-1 and occludin was performed at 6-dpi. Relative to WT neonates, IFNAR−/− neonates had significantly higher whole lung occludin staining and repair gene expression. Together, our data demonstrates IFN-I signaling is extremely pathogenic in the developing lung by disrupting alveolar repair and pulmonary barrier integrity.
期刊介绍:
Mucosal Immunology, the official publication of the Society of Mucosal Immunology (SMI), serves as a forum for both basic and clinical scientists to discuss immunity and inflammation involving mucosal tissues. It covers gastrointestinal, pulmonary, nasopharyngeal, oral, ocular, and genitourinary immunology through original research articles, scholarly reviews, commentaries, editorials, and letters. The journal gives equal consideration to basic, translational, and clinical studies and also serves as a primary communication channel for the SMI governing board and its members, featuring society news, meeting announcements, policy discussions, and job/training opportunities advertisements.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
