{"title":"Gene regulatory network inference during cell fate decisions by perturbation strategies.","authors":"Qing Hu, Xiaoqi Lu, Zhuozhen Xue, Ruiqi Wang","doi":"10.1038/s41540-025-00504-2","DOIUrl":null,"url":null,"abstract":"<p><p>With rapid advances in biological technology and computational approaches, inferring specific gene regulatory networks from data alone during cell fate decisions, including determining direct regulations and their intensities between biomolecules, remains one of the most significant challenges. In this study, we propose a general computational approach based on systematic perturbation, statistical, and differential analyses to infer network topologies and identify network differences during cell fate decisions. For each cell fate state, we first theoretically show how to calculate local response matrices based on perturbation data under systematic perturbation analysis, and we also derive the wild-type (WT) local response matrix for specific ordinary differential equations. To make the inferred network more accurate and eliminate the impact of perturbation degrees, the confidence interval (CI) of local response matrices under multiple perturbations is applied, and the redefined local response matrix is proposed in statistical analysis to determine network topologies across all cell fates. Then in differential analysis, we introduce the concept of relative local response matrix, which enables us to identify critical regulations governing each cell state and dominant cell states associated with specific regulations. The epithelial to mesenchymal transition (EMT) network is chosen as an illustrative example to verify the feasibility of the approach. Largely consistent with experimental observations, the differences of inferred networks at the three cell states can be quantitatively identified. The approach presented here can be also applied to infer other regulatory networks related to cell fate decisions.</p>","PeriodicalId":19345,"journal":{"name":"NPJ Systems Biology and Applications","volume":"11 1","pages":"23"},"PeriodicalIF":3.5000,"publicationDate":"2025-03-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11876352/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Systems Biology and Applications","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41540-025-00504-2","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
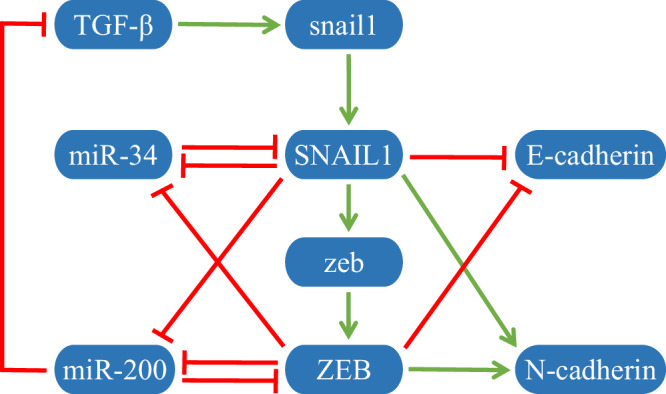
With rapid advances in biological technology and computational approaches, inferring specific gene regulatory networks from data alone during cell fate decisions, including determining direct regulations and their intensities between biomolecules, remains one of the most significant challenges. In this study, we propose a general computational approach based on systematic perturbation, statistical, and differential analyses to infer network topologies and identify network differences during cell fate decisions. For each cell fate state, we first theoretically show how to calculate local response matrices based on perturbation data under systematic perturbation analysis, and we also derive the wild-type (WT) local response matrix for specific ordinary differential equations. To make the inferred network more accurate and eliminate the impact of perturbation degrees, the confidence interval (CI) of local response matrices under multiple perturbations is applied, and the redefined local response matrix is proposed in statistical analysis to determine network topologies across all cell fates. Then in differential analysis, we introduce the concept of relative local response matrix, which enables us to identify critical regulations governing each cell state and dominant cell states associated with specific regulations. The epithelial to mesenchymal transition (EMT) network is chosen as an illustrative example to verify the feasibility of the approach. Largely consistent with experimental observations, the differences of inferred networks at the three cell states can be quantitatively identified. The approach presented here can be also applied to infer other regulatory networks related to cell fate decisions.
期刊介绍:
npj Systems Biology and Applications is an online Open Access journal dedicated to publishing the premier research that takes a systems-oriented approach. The journal aims to provide a forum for the presentation of articles that help define this nascent field, as well as those that apply the advances to wider fields. We encourage studies that integrate, or aid the integration of, data, analyses and insight from molecules to organisms and broader systems. Important areas of interest include not only fundamental biological systems and drug discovery, but also applications to health, medical practice and implementation, big data, biotechnology, food science, human behaviour, broader biological systems and industrial applications of systems biology.
We encourage all approaches, including network biology, application of control theory to biological systems, computational modelling and analysis, comprehensive and/or high-content measurements, theoretical, analytical and computational studies of system-level properties of biological systems and computational/software/data platforms enabling such studies.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
