Rohan Gnanaolivu, Gavin Oliver, Garrett Jenkinson, Emily Blake, Wenan Chen, Nicholas Chia, Eric W Klee, Chen Wang
{"title":"A clinical knowledge graph-based framework to prioritize candidate genes for facilitating diagnosis of Mendelian diseases and rare genetic conditions.","authors":"Rohan Gnanaolivu, Gavin Oliver, Garrett Jenkinson, Emily Blake, Wenan Chen, Nicholas Chia, Eric W Klee, Chen Wang","doi":"10.1186/s12859-025-06096-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Diagnosing Mendelian and rare genetic conditions requires identifying phenotype-associated genetic findings and prioritizing likely disease-causing genes. This task is labor-intensive for molecular and clinical geneticists, who must review extensive literature and databases to link patient phenotypes with causal genotypes. The challenge is further complicated by the large number of genetic variants detected through next-generation sequencing, which impacts both diagnosis timelines and patient care strategies. To address this, in silico methods that prioritize causal genes based on patient-derived phenotypes offer an effective solution, reducing the time involved in diagnostic case reviews and enhancing the efficiency of clinical diagnosis.</p><p><strong>Results: </strong>We developed the phenotype prioritization and analysis for rare diseases (PPAR) to rank genes based on human phenotype ontology (HPO) terms, with the specific goal of aiding the interpretation of genetic testing for Mendelian and rare diseases. PPAR leverages embeddings from a knowledge graph and incorporates knowledge from connections between genes, HPO terms, and gene ontology annotations. When applied on a clinical rare disease cohort and the publicly available deciphering developmental disorders (DDD) dataset. PPAR ranked the causal gene in the top 10 for 27% of cases in the clinical cohort and for 85% of cases in the DDD dataset, outperforming other established HPO-based methods.</p><p><strong>Conclusion: </strong>Our findings demonstrate that PPAR, a method developed from the clinical knowledge graph, effectively ranks causal genes based on patient-derived HPO terms in rare and Mendelian disease contexts. PPAR has shown superior performance compared to other well-established HPO-only methods and provides an efficient, accessible solution for clinical geneticists. The Python-based tool is publicly available at https://github.com/dimi-lab/PPAR , offering a user-friendly platform for gene prioritization.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"82"},"PeriodicalIF":3.3000,"publicationDate":"2025-03-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11908102/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06096-2","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Diagnosing Mendelian and rare genetic conditions requires identifying phenotype-associated genetic findings and prioritizing likely disease-causing genes. This task is labor-intensive for molecular and clinical geneticists, who must review extensive literature and databases to link patient phenotypes with causal genotypes. The challenge is further complicated by the large number of genetic variants detected through next-generation sequencing, which impacts both diagnosis timelines and patient care strategies. To address this, in silico methods that prioritize causal genes based on patient-derived phenotypes offer an effective solution, reducing the time involved in diagnostic case reviews and enhancing the efficiency of clinical diagnosis.
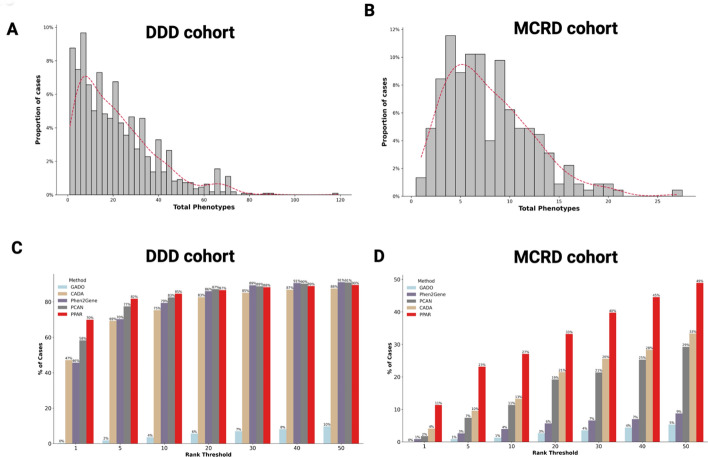
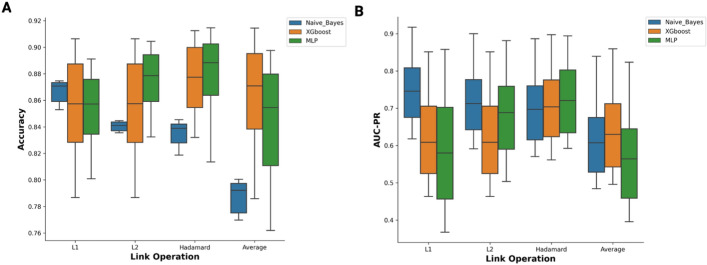
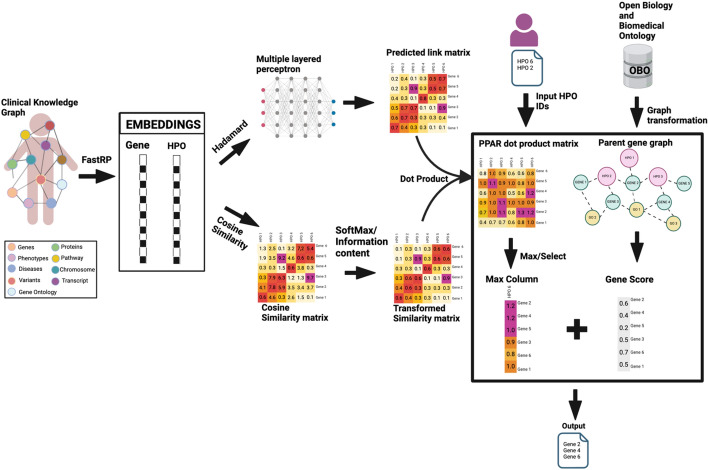
Results: We developed the phenotype prioritization and analysis for rare diseases (PPAR) to rank genes based on human phenotype ontology (HPO) terms, with the specific goal of aiding the interpretation of genetic testing for Mendelian and rare diseases. PPAR leverages embeddings from a knowledge graph and incorporates knowledge from connections between genes, HPO terms, and gene ontology annotations. When applied on a clinical rare disease cohort and the publicly available deciphering developmental disorders (DDD) dataset. PPAR ranked the causal gene in the top 10 for 27% of cases in the clinical cohort and for 85% of cases in the DDD dataset, outperforming other established HPO-based methods.
Conclusion: Our findings demonstrate that PPAR, a method developed from the clinical knowledge graph, effectively ranks causal genes based on patient-derived HPO terms in rare and Mendelian disease contexts. PPAR has shown superior performance compared to other well-established HPO-only methods and provides an efficient, accessible solution for clinical geneticists. The Python-based tool is publicly available at https://github.com/dimi-lab/PPAR , offering a user-friendly platform for gene prioritization.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
