Jeremy M G Leung, Nicolas C Frazee, Alexander Brace, Anthony T Bogetti, Arvind Ramanathan, Lillian T Chong
{"title":"Unsupervised Learning of Progress Coordinates during Weighted Ensemble Simulations: Application to NTL9 Protein Folding.","authors":"Jeremy M G Leung, Nicolas C Frazee, Alexander Brace, Anthony T Bogetti, Arvind Ramanathan, Lillian T Chong","doi":"10.1021/acs.jctc.4c01136","DOIUrl":null,"url":null,"abstract":"<p><p>A major challenge for many rare-event sampling strategies is the identification of progress coordinates that capture the slowest relevant motions. Machine-learning methods that can identify progress coordinates in an unsupervised manner have therefore been of great interest to the simulation community. Here, we developed a general method for identifying progress coordinates \"on-the-fly\" during weighted ensemble (WE) rare-event sampling via deep learning (DL) of outliers among sampled conformations. Our method identifies outliers in a latent space model of the system's sampled conformations that is periodically trained using a convolutional variational autoencoder. As a proof of principle, we applied our DL-enhanced WE method to simulate the NTL9 protein folding process. To enable rapid tests, our simulations propagated discrete-state synthetic molecular dynamics trajectories using a generative, fine-grained Markov state model. Results revealed that our on-the-fly DL of outliers enhanced the efficiency of WE by >3-fold in estimating the folding rate constant. Our efforts are a significant step forward in the unsupervised learning of slow coordinates during rare event sampling.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"3691-3699"},"PeriodicalIF":5.5000,"publicationDate":"2025-04-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11983707/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01136","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/3/19 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
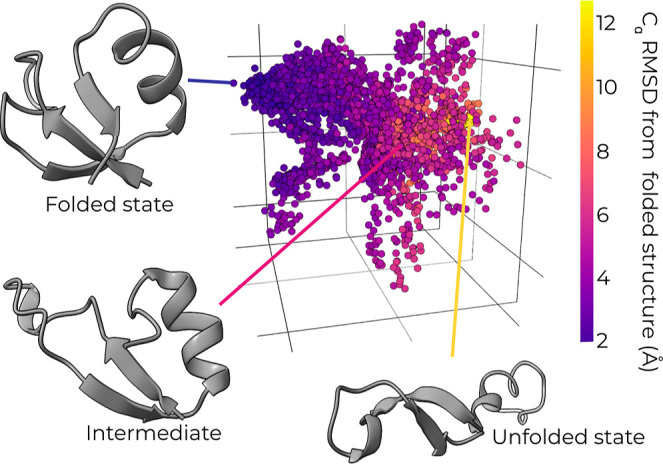
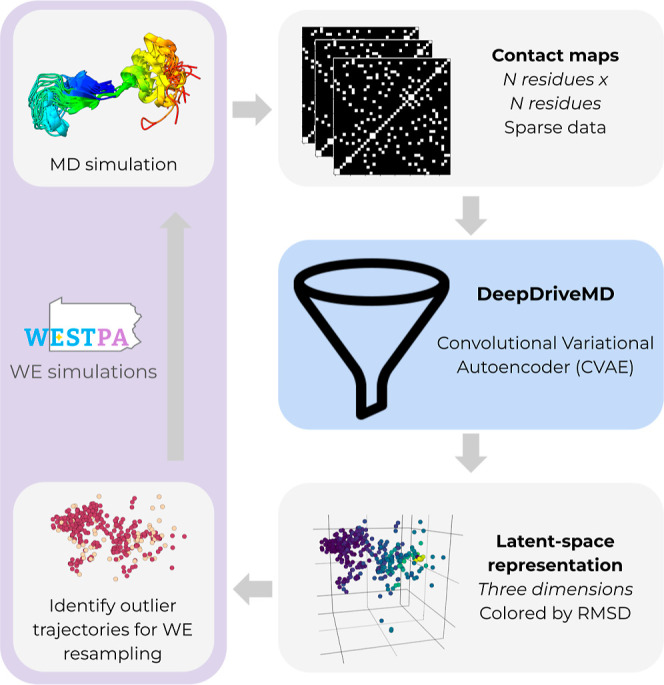
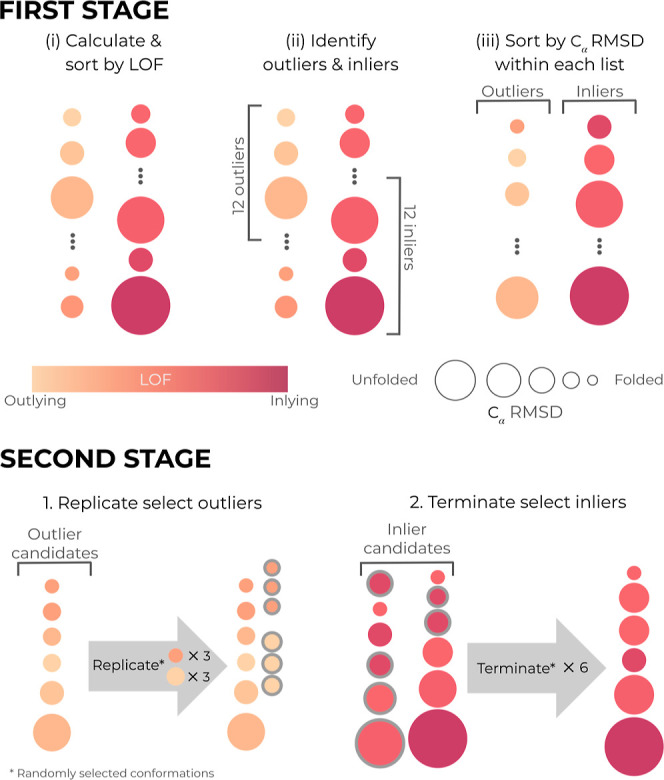
A major challenge for many rare-event sampling strategies is the identification of progress coordinates that capture the slowest relevant motions. Machine-learning methods that can identify progress coordinates in an unsupervised manner have therefore been of great interest to the simulation community. Here, we developed a general method for identifying progress coordinates "on-the-fly" during weighted ensemble (WE) rare-event sampling via deep learning (DL) of outliers among sampled conformations. Our method identifies outliers in a latent space model of the system's sampled conformations that is periodically trained using a convolutional variational autoencoder. As a proof of principle, we applied our DL-enhanced WE method to simulate the NTL9 protein folding process. To enable rapid tests, our simulations propagated discrete-state synthetic molecular dynamics trajectories using a generative, fine-grained Markov state model. Results revealed that our on-the-fly DL of outliers enhanced the efficiency of WE by >3-fold in estimating the folding rate constant. Our efforts are a significant step forward in the unsupervised learning of slow coordinates during rare event sampling.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
