Heli Honkala, Jenni Lahtela, Heli Fox, Massimiliano Gentile, Niklas Pakkasjärvi, Riitta Salonen, Kirmo Wartiovaara, Matti Jauhiainen, Marjo Kestilä
{"title":"Unraveling the disease pathogenesis behind lethal hydrolethalus syndrome revealed multiple changes in molecular and cellular level.","authors":"Heli Honkala, Jenni Lahtela, Heli Fox, Massimiliano Gentile, Niklas Pakkasjärvi, Riitta Salonen, Kirmo Wartiovaara, Matti Jauhiainen, Marjo Kestilä","doi":"10.1186/1755-8417-2-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Hydrolethalus syndrome (HLS) is a severe fetal malformation syndrome characterized by multiple developmental anomalies, including central nervous system (CNS) malformation such as hydrocephaly and absent midline structures of the brain, micrognathia, defective lobation of the lungs and polydactyly. Microscopically, immature cerebral cortex, abnormalities in radial glial cells and hypothalamic hamartoma are among key findings in the CNS of HLS fetuses. HLS is caused by a substitution of aspartic acid by glycine in the HYLS1 protein, whose function was previously unknown.</p><p><strong>Results: </strong>To provide insight into the disease mechanism(s) of this lethal disorder we have studied different aspects of HLS and HYLS1. A genome-wide gene expression analysis indicated several upregulated genes in cell cycle regulatory cascades and in specific signal transduction pathways while many downregulated genes were associated with lipid metabolism. These changes were supported by findings in functional cell biology studies, which revealed an increased cell cycle rate and a decreased amount of apoptosis in HLS neuronal progenitor cells. Also, changes in lipid metabolism gene expression were reflected by a significant increase in the cholesterol levels of HLS liver tissues. In addition, based on our functional studies of HYLS1, we propose that HYLS1 is a transcriptional regulator that shuffles between the cytoplasm and the nucleus, and that when HYLS1 is mutated its function is significantly altered.</p><p><strong>Conclusion: </strong>In this study, we have shown that the HYLS1 mutation has significant consequences in the cellular and tissue levels in HLS fetuses. Based on these results, it can be suggested that HYLS1 is part of the cellular transcriptional regulatory machinery and that the genetic defect has a widespread effect during embryonic and fetal development. These findings add a significant amount of new information to the pathogenesis of HLS and strongly suggest an essential role for HYLS1 in normal fetal development.</p>","PeriodicalId":88084,"journal":{"name":"PathoGenetics","volume":"2 1","pages":"2"},"PeriodicalIF":0.0000,"publicationDate":"2009-04-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1755-8417-2-2","citationCount":"13","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"PathoGenetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1755-8417-2-2","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 13
Abstract
Background: Hydrolethalus syndrome (HLS) is a severe fetal malformation syndrome characterized by multiple developmental anomalies, including central nervous system (CNS) malformation such as hydrocephaly and absent midline structures of the brain, micrognathia, defective lobation of the lungs and polydactyly. Microscopically, immature cerebral cortex, abnormalities in radial glial cells and hypothalamic hamartoma are among key findings in the CNS of HLS fetuses. HLS is caused by a substitution of aspartic acid by glycine in the HYLS1 protein, whose function was previously unknown.
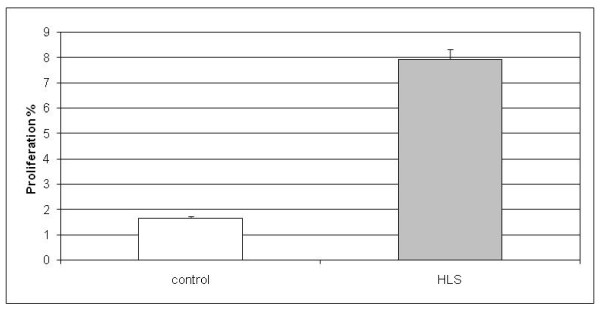
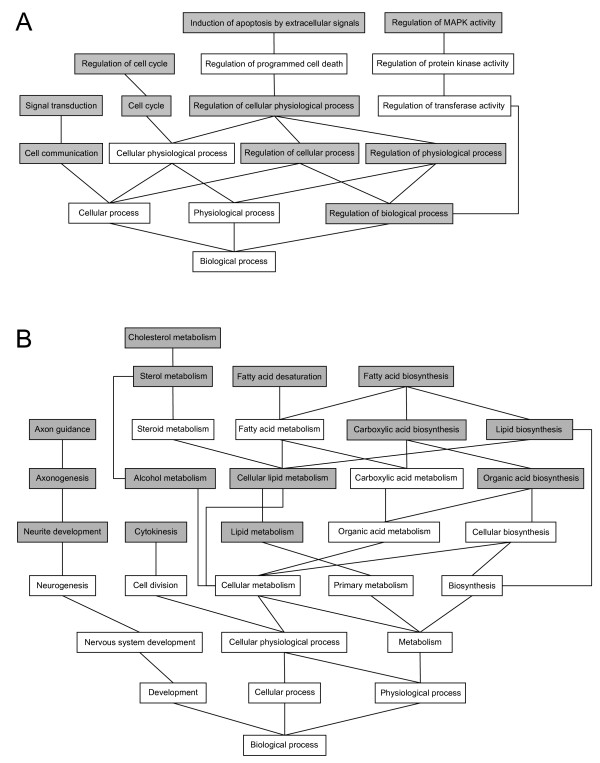
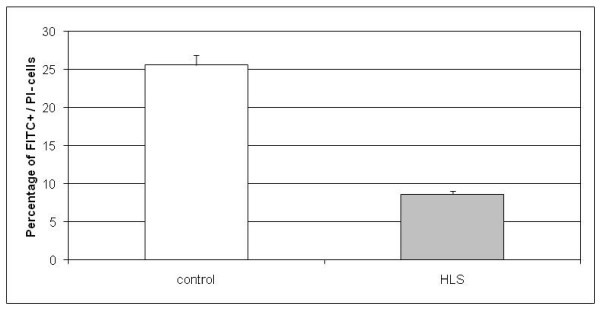
Results: To provide insight into the disease mechanism(s) of this lethal disorder we have studied different aspects of HLS and HYLS1. A genome-wide gene expression analysis indicated several upregulated genes in cell cycle regulatory cascades and in specific signal transduction pathways while many downregulated genes were associated with lipid metabolism. These changes were supported by findings in functional cell biology studies, which revealed an increased cell cycle rate and a decreased amount of apoptosis in HLS neuronal progenitor cells. Also, changes in lipid metabolism gene expression were reflected by a significant increase in the cholesterol levels of HLS liver tissues. In addition, based on our functional studies of HYLS1, we propose that HYLS1 is a transcriptional regulator that shuffles between the cytoplasm and the nucleus, and that when HYLS1 is mutated its function is significantly altered.
Conclusion: In this study, we have shown that the HYLS1 mutation has significant consequences in the cellular and tissue levels in HLS fetuses. Based on these results, it can be suggested that HYLS1 is part of the cellular transcriptional regulatory machinery and that the genetic defect has a widespread effect during embryonic and fetal development. These findings add a significant amount of new information to the pathogenesis of HLS and strongly suggest an essential role for HYLS1 in normal fetal development.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
