Mutations in the nuclear localization sequence of the Aristaless related homeobox; sequestration of mutant ARX with IPO13 disrupts normal subcellular distribution of the transcription factor and retards cell division.
Cheryl Shoubridge, May Huey Tan, Tod Fullston, Desiree Cloosterman, David Coman, George McGillivray, Grazia M Mancini, Tjitske Kleefstra, Jozef Gécz
{"title":"Mutations in the nuclear localization sequence of the Aristaless related homeobox; sequestration of mutant ARX with IPO13 disrupts normal subcellular distribution of the transcription factor and retards cell division.","authors":"Cheryl Shoubridge, May Huey Tan, Tod Fullston, Desiree Cloosterman, David Coman, George McGillivray, Grazia M Mancini, Tjitske Kleefstra, Jozef Gécz","doi":"10.1186/1755-8417-3-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Aristaless related homeobox (ARX) is a paired-type homeobox gene. ARX function is frequently affected by naturally occurring mutations. Nonsense mutations, polyalanine tract expansions and missense mutations in ARX cause a range of intellectual disability and epilepsy phenotypes with or without additional features including hand dystonia, lissencephaly, autism or dysarthria. Severe malformation phenotypes, such as X-linked lissencephaly with ambiguous genitalia (XLAG), are frequently observed in individuals with protein truncating or missense mutations clustered in the highly conserved paired-type homeodomain.</p><p><strong>Results: </strong>We have identified two novel point mutations in the R379 residue of the ARX homeodomain; c.1135C>A, p.R379S in a patient with infantile spasms and intellectual disability and c.1136G>T, p.R379L in a patient with XLAG. We investigated these and other missense mutations (R332P, R332H, R332C, T333N: associated with XLAG and Proud syndrome) predicted to affect the nuclear localisation sequences (NLS) flanking either end of the ARX homeodomain. The NLS regions are required for correct nuclear import facilitated by Importin 13 (IPO13). We demonstrate that missense mutations in either the N- or C-terminal NLS regions of the homeodomain cause significant disruption to nuclear localisation of the ARX protein in vitro. Surprisingly, none of these mutations abolished the binding of ARX to IPO13. This was confirmed by co-immunoprecipitation and immmuno fluorescence studies. Instead, tagged and endogenous IPO13 remained bound to the mutant ARX proteins, even in the RanGTP rich nuclear environment. We also identify the microtubule protein TUBA1A as a novel interacting protein for ARX and show cells expressing mutant ARX protein accumulate in mitosis, indicating normal cell division may be disrupted.</p><p><strong>Conclusions: </strong>We show that the most likely, common pathogenic mechanism of the missense mutations in NLS regions of the ARX homeodomain is inadequate accumulation and distribution of the ARX transcription factor within the nucleus due to sequestration of ARX with IPO13.</p>","PeriodicalId":88084,"journal":{"name":"PathoGenetics","volume":"3 ","pages":"1"},"PeriodicalIF":0.0000,"publicationDate":"2010-01-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1755-8417-3-1","citationCount":"37","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"PathoGenetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1755-8417-3-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 37
Abstract
Background: Aristaless related homeobox (ARX) is a paired-type homeobox gene. ARX function is frequently affected by naturally occurring mutations. Nonsense mutations, polyalanine tract expansions and missense mutations in ARX cause a range of intellectual disability and epilepsy phenotypes with or without additional features including hand dystonia, lissencephaly, autism or dysarthria. Severe malformation phenotypes, such as X-linked lissencephaly with ambiguous genitalia (XLAG), are frequently observed in individuals with protein truncating or missense mutations clustered in the highly conserved paired-type homeodomain.
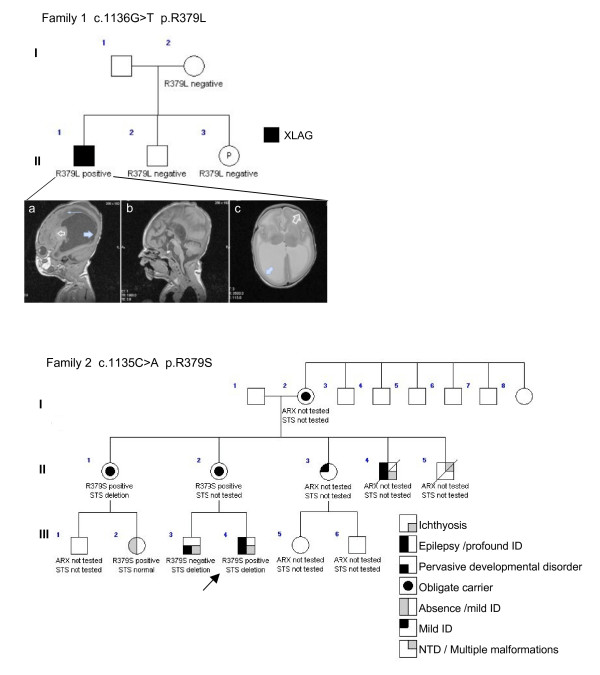
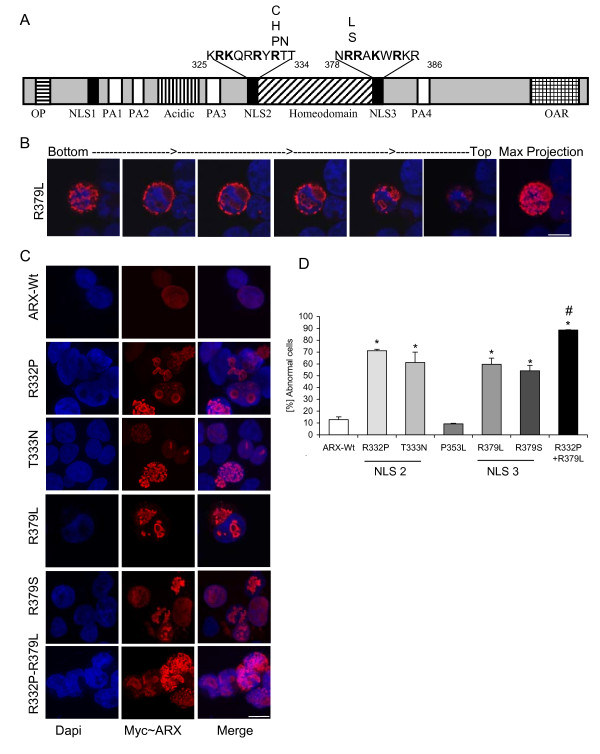
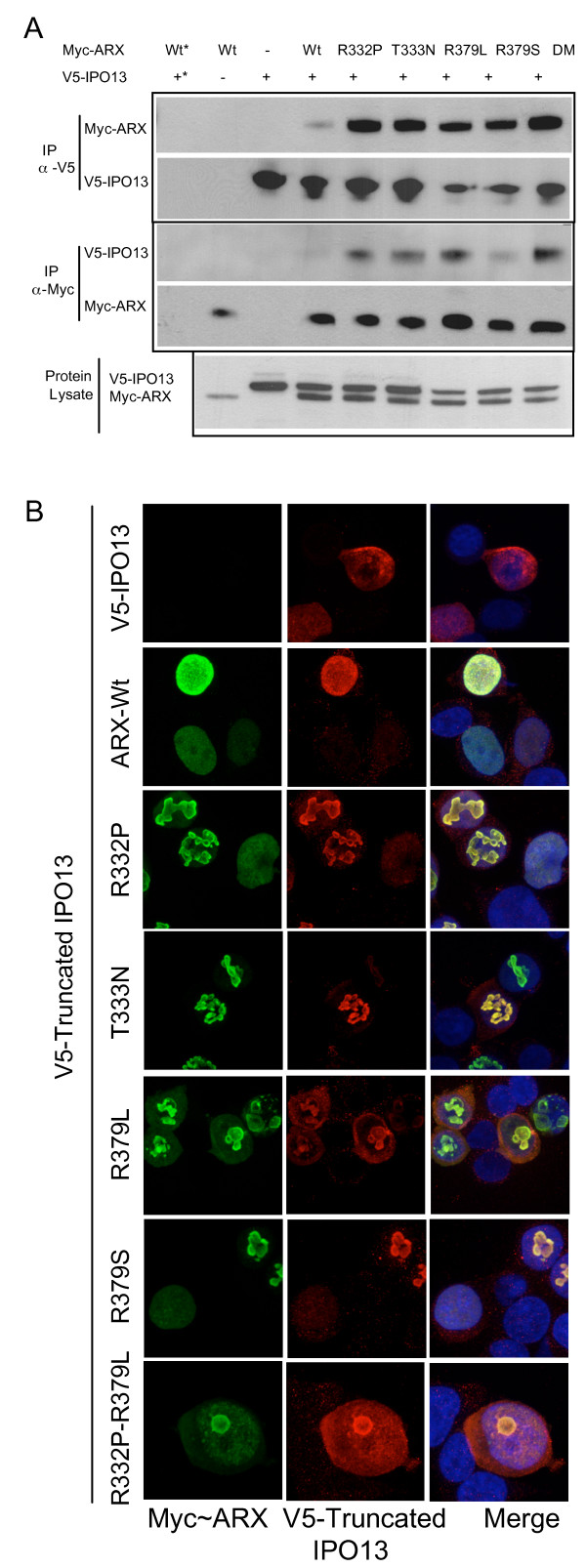
Results: We have identified two novel point mutations in the R379 residue of the ARX homeodomain; c.1135C>A, p.R379S in a patient with infantile spasms and intellectual disability and c.1136G>T, p.R379L in a patient with XLAG. We investigated these and other missense mutations (R332P, R332H, R332C, T333N: associated with XLAG and Proud syndrome) predicted to affect the nuclear localisation sequences (NLS) flanking either end of the ARX homeodomain. The NLS regions are required for correct nuclear import facilitated by Importin 13 (IPO13). We demonstrate that missense mutations in either the N- or C-terminal NLS regions of the homeodomain cause significant disruption to nuclear localisation of the ARX protein in vitro. Surprisingly, none of these mutations abolished the binding of ARX to IPO13. This was confirmed by co-immunoprecipitation and immmuno fluorescence studies. Instead, tagged and endogenous IPO13 remained bound to the mutant ARX proteins, even in the RanGTP rich nuclear environment. We also identify the microtubule protein TUBA1A as a novel interacting protein for ARX and show cells expressing mutant ARX protein accumulate in mitosis, indicating normal cell division may be disrupted.
Conclusions: We show that the most likely, common pathogenic mechanism of the missense mutations in NLS regions of the ARX homeodomain is inadequate accumulation and distribution of the ARX transcription factor within the nucleus due to sequestration of ARX with IPO13.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
