Luiz Augusto G Silva, Luis Antonio B Kowada, Noraí Romeu Rocco, Maria Emília M T Walter
{"title":"A new 1.375-approximation algorithm for sorting by transpositions.","authors":"Luiz Augusto G Silva, Luis Antonio B Kowada, Noraí Romeu Rocco, Maria Emília M T Walter","doi":"10.1186/s13015-022-00205-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>SORTING BY TRANSPOSITIONS (SBT) is a classical problem in genome rearrangements. In 2012, SBT was proven to be [Formula: see text]-hard and the best approximation algorithm with a 1.375 ratio was proposed in 2006 by Elias and Hartman (EH algorithm). Their algorithm employs simplification, a technique used to transform an input permutation [Formula: see text] into a simple permutation [Formula: see text], presumably easier to handle with. The permutation [Formula: see text] is obtained by inserting new symbols into [Formula: see text] in a way that the lower bound of the transposition distance of [Formula: see text] is kept on [Formula: see text]. The simplification is guaranteed to keep the lower bound, not the transposition distance. A sequence of operations sorting [Formula: see text] can be mimicked to sort [Formula: see text].</p><p><strong>Results and conclusions: </strong>First, using an algebraic approach, we propose a new upper bound for the transposition distance, which holds for all [Formula: see text]. Next, motivated by a problem identified in the EH algorithm, which causes it, in scenarios involving how the input permutation is simplified, to require one extra transposition above the 1.375-approximation ratio, we propose a new approximation algorithm to solve SBT ensuring the 1.375-approximation ratio for all [Formula: see text]. We implemented our algorithm and EH's. Regarding the implementation of the EH algorithm, two other issues were identified and needed to be fixed. We tested both algorithms against all permutations of size n, [Formula: see text]. The results show that the EH algorithm exceeds the approximation ratio of 1.375 for permutations with a size greater than 7. The percentage of computed distances that are equal to transposition distance, computed by the implemented algorithms are also compared with others available in the literature. Finally, we investigate the performance of both implementations on longer permutations of maximum length 500. From the experiments, we conclude that maximum and the average distances computed by our algorithm are a little better than the ones computed by the EH algorithm and the running times of both algorithms are similar, despite the time complexity of our algorithm being higher.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":" ","pages":"1"},"PeriodicalIF":1.7000,"publicationDate":"2022-01-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8760837/pdf/","citationCount":"3","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-022-00205-z","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 3
Abstract
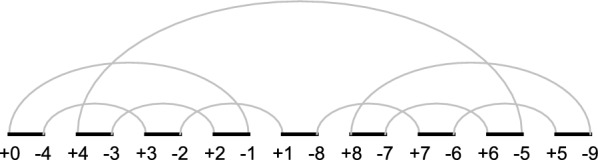
Background: SORTING BY TRANSPOSITIONS (SBT) is a classical problem in genome rearrangements. In 2012, SBT was proven to be [Formula: see text]-hard and the best approximation algorithm with a 1.375 ratio was proposed in 2006 by Elias and Hartman (EH algorithm). Their algorithm employs simplification, a technique used to transform an input permutation [Formula: see text] into a simple permutation [Formula: see text], presumably easier to handle with. The permutation [Formula: see text] is obtained by inserting new symbols into [Formula: see text] in a way that the lower bound of the transposition distance of [Formula: see text] is kept on [Formula: see text]. The simplification is guaranteed to keep the lower bound, not the transposition distance. A sequence of operations sorting [Formula: see text] can be mimicked to sort [Formula: see text].
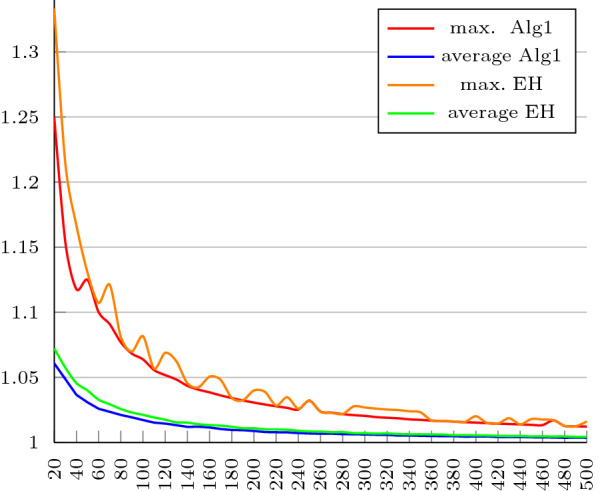
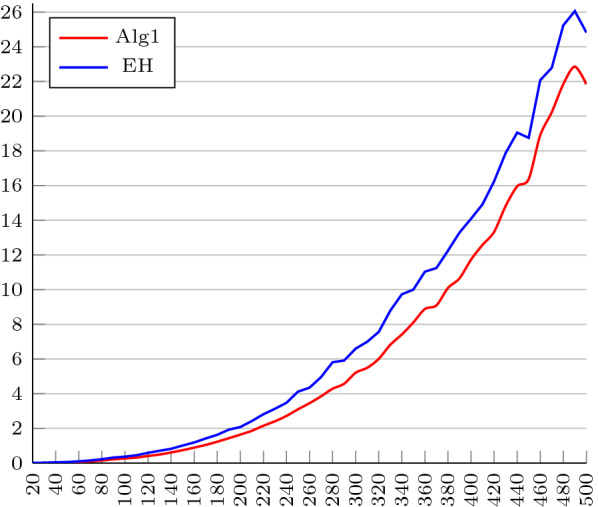
Results and conclusions: First, using an algebraic approach, we propose a new upper bound for the transposition distance, which holds for all [Formula: see text]. Next, motivated by a problem identified in the EH algorithm, which causes it, in scenarios involving how the input permutation is simplified, to require one extra transposition above the 1.375-approximation ratio, we propose a new approximation algorithm to solve SBT ensuring the 1.375-approximation ratio for all [Formula: see text]. We implemented our algorithm and EH's. Regarding the implementation of the EH algorithm, two other issues were identified and needed to be fixed. We tested both algorithms against all permutations of size n, [Formula: see text]. The results show that the EH algorithm exceeds the approximation ratio of 1.375 for permutations with a size greater than 7. The percentage of computed distances that are equal to transposition distance, computed by the implemented algorithms are also compared with others available in the literature. Finally, we investigate the performance of both implementations on longer permutations of maximum length 500. From the experiments, we conclude that maximum and the average distances computed by our algorithm are a little better than the ones computed by the EH algorithm and the running times of both algorithms are similar, despite the time complexity of our algorithm being higher.
背景:转位排序(SBT)是基因组重排中的经典问题。2012年,SBT被证明为[公式:见文]-hard, 2006年,Elias和Hartman提出了1.375的最佳近似算法(EH算法)。他们的算法采用简化,一种将输入排列[公式:见文本]转换为简单排列[公式:见文本]的技术,想必更容易处理。通过在[Formula: see text]中插入新的符号,使[Formula: see text]的换位距离下界保持在[Formula: see text]上,得到[Formula: see text]的排列。简化保证了保留下界,而不是移位距离。排序的操作序列[公式:见文本]可以模拟排序[公式:见文本]。结果和结论:首先,使用代数方法,我们提出了一个新的移位距离上界,该上界适用于所有[公式:见文本]。接下来,在EH算法中发现的一个问题的激励下,在涉及如何简化输入排列的场景中,它需要在1.375近似比之上额外进行一次换位,我们提出了一种新的近似算法来解决SBT,确保所有的近似比都是1.375[公式:见文本]。我们实现了我们的算法和EH。关于EH算法的实现,还发现了另外两个需要解决的问题。我们针对大小为n的所有排列测试了这两种算法,[公式:见文本]。结果表明,EH算法对于大小大于7的排列超过了1.375的近似比。由实现的算法计算的与换位距离相等的计算距离的百分比也与文献中其他可用的算法进行了比较。最后,我们研究了两种实现在最大长度为500的更长的排列上的性能。实验结果表明,尽管算法的时间复杂度较高,但算法计算的最大距离和平均距离略优于EH算法,两种算法的运行时间相似。
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
