Zi-Jun Quan , Si-Ang Li , Zhi-Xue Yang , Juan-Juan Zhao , Guo-Hua Li , Feng Zhang , Wei Wen , Tao Cheng , Xiao-Bing Zhang
{"title":"GREPore-seq: A Robust Workflow to Detect Changes After Gene Editing Through Long-range PCR and Nanopore Sequencing","authors":"Zi-Jun Quan , Si-Ang Li , Zhi-Xue Yang , Juan-Juan Zhao , Guo-Hua Li , Feng Zhang , Wei Wen , Tao Cheng , Xiao-Bing Zhang","doi":"10.1016/j.gpb.2022.06.002","DOIUrl":null,"url":null,"abstract":"<div><div>To achieve the enormous potential of gene-editing technology in clinical therapies, one needs to evaluate both the on-target efficiency and unintended editing consequences comprehensively. However, there is a lack of a pipelined, large-scale, and economical workflow for detecting genome editing outcomes, in particular insertion or deletion of a large fragment. Here, we describe an approach for efficient and accurate detection of multiple <strong>genetic changes</strong> after <strong>CRISPR/Cas9</strong> editing by pooled <strong>nanopore sequencing</strong> of barcoded <strong>long-range PCR</strong> products. Recognizing the high error rates of Oxford nanopore sequencing, we developed a novel pipeline to capture the barcoded sequences by grepping reads of nanopore amplicon sequencing (<strong>GREPore-seq</strong>). GREPore-seq can assess nonhomologous end-joining (NHEJ)-mediated double-stranded oligodeoxynucleotide (dsODN) insertions with comparable accuracy to Illumina next-generation sequencing (NGS). GREPore-seq also reveals a full spectrum of homology-directed repair (HDR)-mediated large gene knock-in, correlating well with the fluorescence-activated cell sorting (FACS) analysis results. Of note, we discovered low-level fragmented and full-length plasmid backbone insertion at the CRISPR cutting site. Therefore, we have established a practical workflow to evaluate various genetic changes, including quantifying insertions of short dsODNs, knock-ins of long pieces, plasmid insertions, and large fragment deletions after CRISPR/Cas9-mediated editing. GREPore-seq is freely available at GitHub (<span><span>https://github.com/lisiang/GREPore-seq</span><svg><path></path></svg></span>) and the National Genomics Data Center (NGDC) BioCode (<span><span>https://ngdc.cncb.ac.cn/biocode/tools/BT007293</span><svg><path></path></svg></span>).</div></div>","PeriodicalId":12528,"journal":{"name":"Genomics, Proteomics & Bioinformatics","volume":"21 6","pages":"Pages 1221-1236"},"PeriodicalIF":7.9000,"publicationDate":"2023-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11082256/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genomics, Proteomics & Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S167202292200078X","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
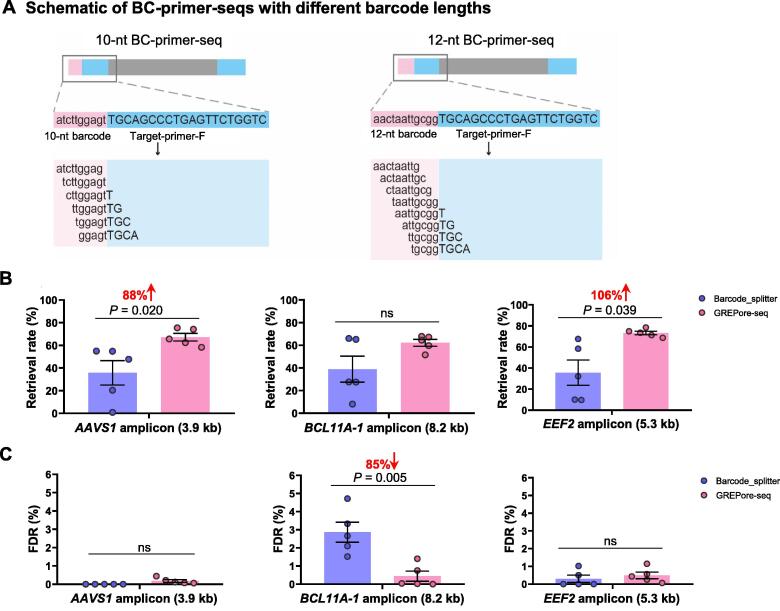
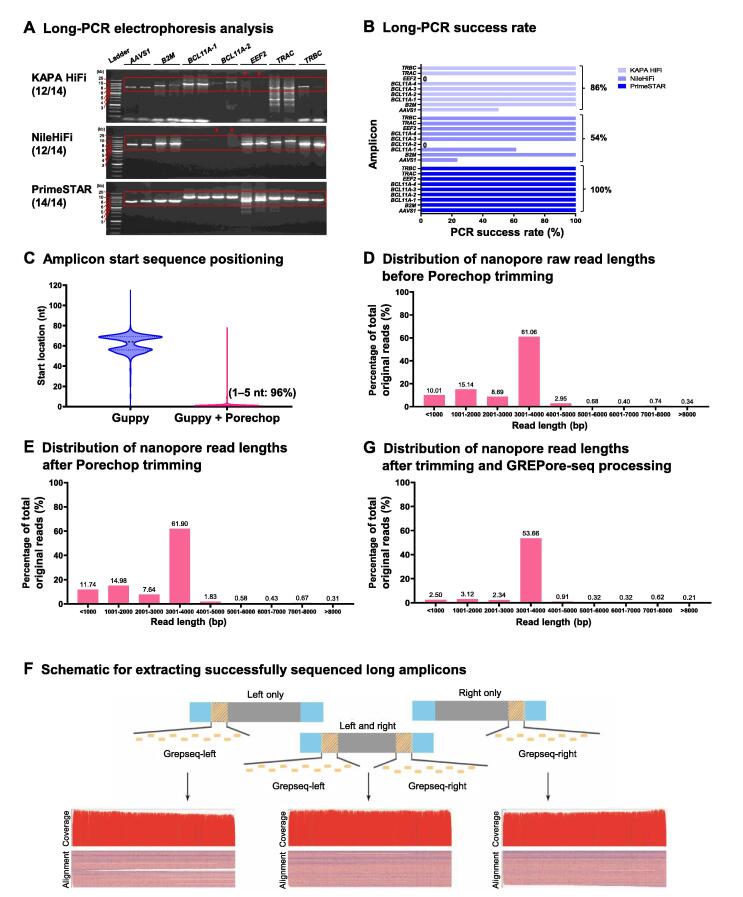
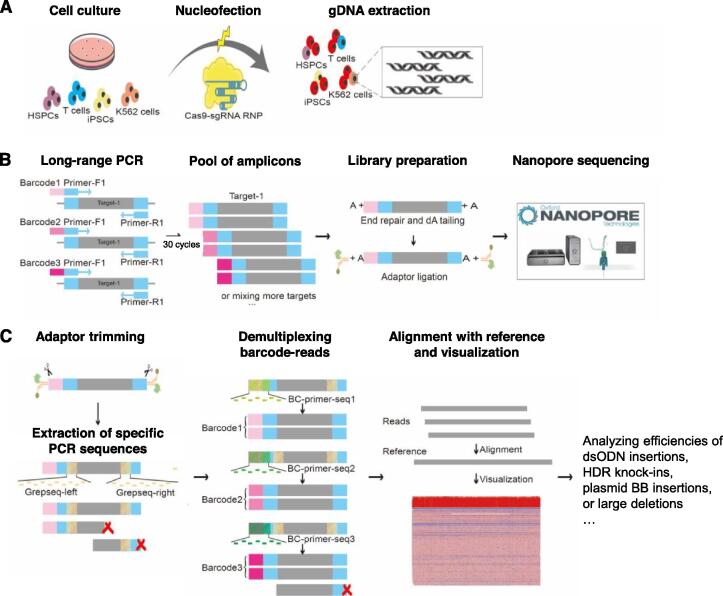
To achieve the enormous potential of gene-editing technology in clinical therapies, one needs to evaluate both the on-target efficiency and unintended editing consequences comprehensively. However, there is a lack of a pipelined, large-scale, and economical workflow for detecting genome editing outcomes, in particular insertion or deletion of a large fragment. Here, we describe an approach for efficient and accurate detection of multiple genetic changes after CRISPR/Cas9 editing by pooled nanopore sequencing of barcoded long-range PCR products. Recognizing the high error rates of Oxford nanopore sequencing, we developed a novel pipeline to capture the barcoded sequences by grepping reads of nanopore amplicon sequencing (GREPore-seq). GREPore-seq can assess nonhomologous end-joining (NHEJ)-mediated double-stranded oligodeoxynucleotide (dsODN) insertions with comparable accuracy to Illumina next-generation sequencing (NGS). GREPore-seq also reveals a full spectrum of homology-directed repair (HDR)-mediated large gene knock-in, correlating well with the fluorescence-activated cell sorting (FACS) analysis results. Of note, we discovered low-level fragmented and full-length plasmid backbone insertion at the CRISPR cutting site. Therefore, we have established a practical workflow to evaluate various genetic changes, including quantifying insertions of short dsODNs, knock-ins of long pieces, plasmid insertions, and large fragment deletions after CRISPR/Cas9-mediated editing. GREPore-seq is freely available at GitHub (https://github.com/lisiang/GREPore-seq) and the National Genomics Data Center (NGDC) BioCode (https://ngdc.cncb.ac.cn/biocode/tools/BT007293).
期刊介绍:
Genomics, Proteomics and Bioinformatics (GPB) is the official journal of the Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation and Genetics Society of China. It aims to disseminate new developments in the field of omics and bioinformatics, publish high-quality discoveries quickly, and promote open access and online publication. GPB welcomes submissions in all areas of life science, biology, and biomedicine, with a focus on large data acquisition, analysis, and curation. Manuscripts covering omics and related bioinformatics topics are particularly encouraged. GPB is indexed/abstracted by PubMed/MEDLINE, PubMed Central, Scopus, BIOSIS Previews, Chemical Abstracts, CSCD, among others.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
