Lars L. J. van der Burg, Liesbeth C. de Wreede, Henning Baldauf, Jürgen Sauter, Johannes Schetelig, Hein Putter, Stefan Böhringer
{"title":"Haplotype reconstruction for genetically complex regions with ambiguous genotype calls: Illustration by the KIR gene region","authors":"Lars L. J. van der Burg, Liesbeth C. de Wreede, Henning Baldauf, Jürgen Sauter, Johannes Schetelig, Hein Putter, Stefan Böhringer","doi":"10.1002/gepi.22538","DOIUrl":null,"url":null,"abstract":"<p>Advances in DNA sequencing technologies have enabled genotyping of complex genetic regions exhibiting copy number variation and high allelic diversity, yet it is impossible to derive exact genotypes in all cases, often resulting in ambiguous genotype calls, that is, partially missing data. An example of such a gene region is the killer-cell immunoglobulin-like receptor (<i>KIR</i>) genes. These genes are of special interest in the context of allogeneic hematopoietic stem cell transplantation. For such complex gene regions, current haplotype reconstruction methods are not feasible as they cannot cope with the complexity of the data. We present an expectation–maximization (EM)-algorithm to estimate haplotype frequencies (HTFs) which deals with the missing data components, and takes into account linkage disequilibrium (LD) between genes. To cope with the exponential increase in the number of haplotypes as genes are added, we add three components to a standard EM-algorithm implementation. First, reconstruction is performed iteratively, adding one gene at a time. Second, after each step, haplotypes with frequencies below a threshold are collapsed in a rare haplotype group. Third, the HTF of the rare haplotype group is profiled in subsequent iterations to improve estimates. A simulation study evaluates the effect of combining information of multiple genes on the estimates of these frequencies. We show that estimated HTFs are approximately unbiased. Our simulation study shows that the EM-algorithm is able to combine information from multiple genes when LD is high, whereas increased ambiguity levels increase bias. Linear regression models based on this EM, show that a large number of haplotypes can be problematic for unbiased effect size estimation and that models need to be sparse. In a real data analysis of <i>KIR</i> genotypes, we compare HTFs to those obtained in an independent study. Our new EM-algorithm-based method is the first to account for the full genetic architecture of complex gene regions, such as the <i>KIR</i> gene region. This algorithm can handle the numerous observed ambiguities, and allows for the collapsing of haplotypes to perform implicit dimension reduction. Combining information from multiple genes improves haplotype reconstruction.</p>","PeriodicalId":12710,"journal":{"name":"Genetic Epidemiology","volume":"48 1","pages":"3-26"},"PeriodicalIF":3.8000,"publicationDate":"2023-10-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/gepi.22538","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Genetic Epidemiology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/gepi.22538","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
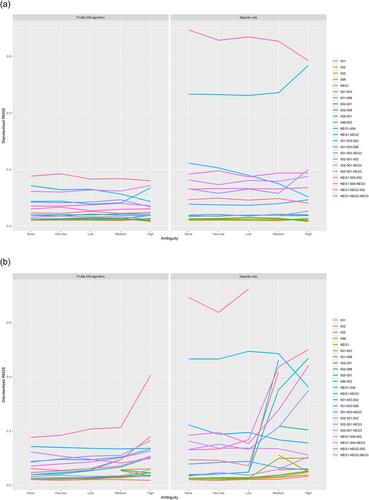
Advances in DNA sequencing technologies have enabled genotyping of complex genetic regions exhibiting copy number variation and high allelic diversity, yet it is impossible to derive exact genotypes in all cases, often resulting in ambiguous genotype calls, that is, partially missing data. An example of such a gene region is the killer-cell immunoglobulin-like receptor (KIR) genes. These genes are of special interest in the context of allogeneic hematopoietic stem cell transplantation. For such complex gene regions, current haplotype reconstruction methods are not feasible as they cannot cope with the complexity of the data. We present an expectation–maximization (EM)-algorithm to estimate haplotype frequencies (HTFs) which deals with the missing data components, and takes into account linkage disequilibrium (LD) between genes. To cope with the exponential increase in the number of haplotypes as genes are added, we add three components to a standard EM-algorithm implementation. First, reconstruction is performed iteratively, adding one gene at a time. Second, after each step, haplotypes with frequencies below a threshold are collapsed in a rare haplotype group. Third, the HTF of the rare haplotype group is profiled in subsequent iterations to improve estimates. A simulation study evaluates the effect of combining information of multiple genes on the estimates of these frequencies. We show that estimated HTFs are approximately unbiased. Our simulation study shows that the EM-algorithm is able to combine information from multiple genes when LD is high, whereas increased ambiguity levels increase bias. Linear regression models based on this EM, show that a large number of haplotypes can be problematic for unbiased effect size estimation and that models need to be sparse. In a real data analysis of KIR genotypes, we compare HTFs to those obtained in an independent study. Our new EM-algorithm-based method is the first to account for the full genetic architecture of complex gene regions, such as the KIR gene region. This algorithm can handle the numerous observed ambiguities, and allows for the collapsing of haplotypes to perform implicit dimension reduction. Combining information from multiple genes improves haplotype reconstruction.
期刊介绍:
Genetic Epidemiology is a peer-reviewed journal for discussion of research on the genetic causes of the distribution of human traits in families and populations. Emphasis is placed on the relative contribution of genetic and environmental factors to human disease as revealed by genetic, epidemiological, and biologic investigations.
Genetic Epidemiology primarily publishes papers in statistical genetics, a research field that is primarily concerned with development of statistical, bioinformatical, and computational models for analyzing genetic data. Incorporation of underlying biology and population genetics into conceptual models is favored. The Journal seeks original articles comprising either applied research or innovative statistical, mathematical, computational, or genomic methodologies that advance studies in genetic epidemiology. Other types of reports are encouraged, such as letters to the editor, topic reviews, and perspectives from other fields of research that will likely enrich the field of genetic epidemiology.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
